
International Journal of
Reproductive Biomedicine
Tue, Aug 4, 2026
[Archive]
Volume 1, Issue 1 (1-2003)
IJRM 2003, 1(1): 12-15 |
Back to browse issues page

![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Movassagh-Pour A A, Salehnia M, Pourfatollah A A, Moazzeni S M. The Effect of Murine Leukemia Inhibitory Factor on In Vitro Differentiation of Mouse Embryonic Stem Cells. IJRM 2003; 1 (1) :12-15
URL: http://ijrm.ir/article-1-1-en.html
URL: http://ijrm.ir/article-1-1-en.html


Full-Text [PDF 170 kb]
(764 Downloads)
| Abstract (HTML) (4002 Views)
The mouse embryos at early developmental stages contain a population of cells, which are totipotential and have the ability to differentiate to somatic and germ cells (Smith 1992). Undifferentiated ICM from several mammalian blastocysts have been isolated and cultured in vitro (Pera et al., 2000; Reubinoff et al., 2000;Thomson and Marshal, 1998; Bongso et al., 1994). If these totipotent ESc are made to proliferate without differentiation could have several usages such as cell therapy, production of transgenic animals, as a model for studying the mechanism of embryo development, gene targeting and gene transfer (Odorico et al., 2001; Rathjeh et al., 1998).
The ESc have some unique properties. These cells are immortal and under influence of some factors which may differentiate into several cell lines such as endothelial, neuroepithelial, hematopoietic, and muscle cells (Smith, 1998). At first ES cell lines were obtained from rabbit embryos by Cole et al. (1965). Later, Evans and Kaufman (1981) and Martin (1981) reported their success in isolation of ESc line from mouse embryos. They cultured mouse ESc on the primary fibroblast feeder layer. Some investigators believed that believed that co-culture of ESc with feeder layers of mitotically inactivated is not essential; however, in the presence of LIF and 2 mercaptoethanol, the ESc sustain cell survival and were undifferentiated (Smith et al., 1988; Smith and Hooper 1987).
The objective of this experimental study was to isolate the ESc from NMRI mouse blastocyst stage and compare the efficiency of LIF to inhibiting the differentiation of mouse ES.
Materials and Methods
Collection of embryo
Adult NMRI mice were maintained on a 12 h light and 12 h dark cycle. Embryos were collected by flushing the uterine horns4 days after ovarian hyper stimulation using 10 IU injection of hMG, followed 48h later with another injection of 10 IU of hCG and natural mating. The presence of vaginal plug was examined on the morning of day one. The embryos were cultured in DMEM medium supplemented with 10% FBS for 1-2 days to allow hatching.
Embryo culture and isolation of ICM colony
The hatched blastocysts were cultured for formation of ICM colonies and their attachment to the bottom of plate. The ICM colonies were collected using the blunt heat sealed end of Pasteur pipette and transferred to a drop of 0.25% trypsin 0.02% EDTA under mineral oil.These were then incubated for 3-4 min at 37ºC, 5% Co2 and 95% air. After that the cells were separated using very fine Pasteur pipette filled with DMEM medium and washing the cells several times with fresh medium. The isolated cells were transferred to 96 well dishes containing DMEM supplemented with 10% FBS and 10% newborn calf serum (NCS), 0.1 mM β mercaptoethanol and 1000 U/ml LIF. Cultures were incubated for two weeks at 37ºC in water saturated atmosphere and 5% Co2. Medium was renewed after two or three days. Each well was examined every day up to 14 days. The colonies having typical EscMorphology were subcultured. The sub culturing method was similar to the previous procedure.

Results
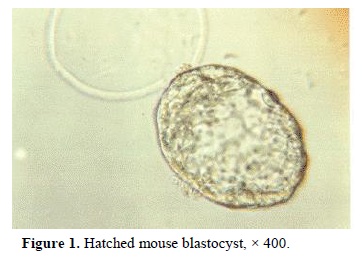
The summary of embryo development and formation of ICM and ES colonies are presented in Table I. 78% of embryos were hatched following incubation in DMEM for 12-14 h (Figure 1). After hatching, there were two morphologically distinct cell types, ICM colonies and trophectoderm. Later, cells
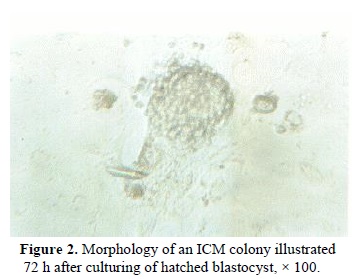
were attached to the bottom of plate as a monolayer. The ICM cell colonies were small cells with a large nucleus and minimal cytoplasm (Figure 2). These cells were tightly packed and before trypsinizing were
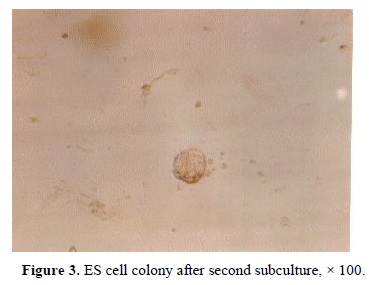
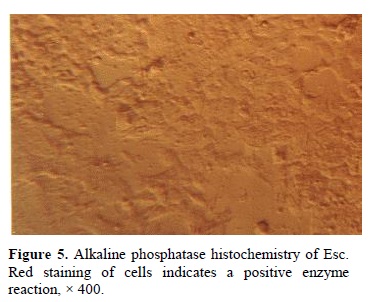
were tightly packed and before trypsinizing were difficult to recognize individual cells. The morphology of ICM colonies in the first, second and third subcultures were similar (Figure 3). However, they formed monolayer on the plate following forth subculture (Figure 4). 89% of the hatched embryos formed ICM colonies; however, only 10% of ICM colonies could produce ESc. Red staining of the cells after alkaline phosphatase histochemistry, indicated a positive enzyme reaction, which demonstrated in Figure 5.

Discussion
The main purpose of this study was to establish the isolation and production of NMRI mouse ES cells. Our results showed that the ICM colonies that were obtained from hatched NMRI mouse blastocyst could produce ES cells. After some sub culturing, the morphology of those cells were similar to the other ES cell lines, which have been obtained from another strain of mammals such as human (Bongso et al.,1994; Thomson et al., 1995; Doetschman et al., 1988). The alkaline phosphatase reaction showed that these cells became undifferentiated after some subculturing.
Our result also demonstrated that LIF in the absence of feeder layer is efficient in preventing the differentiation of ES cells. In contrast, Anderson (1992) showed an unsuccessful experiments for usage of murine and human LIF to assist isolation of ESc. Therefore, it appears that the action of LIF on ICM cells may be species as well as strain dependent.
In conclusion, the results showed that a highly pluripotent ESc can be derived from blastocyst of NMRI mice. It was also demonstrated that these cells could be differentiated into GM-CSF hematopoietic colonies (Movassagh Pour et al., 2003). Thus, this technique could be effective to produce ESc which may be used in the future reproductive studies.
Acknowledgement
The authors would like to thank Mr M. Solaymani for technical assistance. This study was funded by Iran Blood Transfusion Organization.
Full-Text: (490 Views)
Introduction
The mouse embryos at early developmental stages contain a population of cells, which are totipotential and have the ability to differentiate to somatic and germ cells (Smith 1992). Undifferentiated ICM from several mammalian blastocysts have been isolated and cultured in vitro (Pera et al., 2000; Reubinoff et al., 2000;Thomson and Marshal, 1998; Bongso et al., 1994). If these totipotent ESc are made to proliferate without differentiation could have several usages such as cell therapy, production of transgenic animals, as a model for studying the mechanism of embryo development, gene targeting and gene transfer (Odorico et al., 2001; Rathjeh et al., 1998).
The ESc have some unique properties. These cells are immortal and under influence of some factors which may differentiate into several cell lines such as endothelial, neuroepithelial, hematopoietic, and muscle cells (Smith, 1998). At first ES cell lines were obtained from rabbit embryos by Cole et al. (1965). Later, Evans and Kaufman (1981) and Martin (1981) reported their success in isolation of ESc line from mouse embryos. They cultured mouse ESc on the primary fibroblast feeder layer. Some investigators believed that believed that co-culture of ESc with feeder layers of mitotically inactivated is not essential; however, in the presence of LIF and 2 mercaptoethanol, the ESc sustain cell survival and were undifferentiated (Smith et al., 1988; Smith and Hooper 1987).
The objective of this experimental study was to isolate the ESc from NMRI mouse blastocyst stage and compare the efficiency of LIF to inhibiting the differentiation of mouse ES.
Materials and Methods
Collection of embryo
Adult NMRI mice were maintained on a 12 h light and 12 h dark cycle. Embryos were collected by flushing the uterine horns4 days after ovarian hyper stimulation using 10 IU injection of hMG, followed 48h later with another injection of 10 IU of hCG and natural mating. The presence of vaginal plug was examined on the morning of day one. The embryos were cultured in DMEM medium supplemented with 10% FBS for 1-2 days to allow hatching.
Embryo culture and isolation of ICM colony
The hatched blastocysts were cultured for formation of ICM colonies and their attachment to the bottom of plate. The ICM colonies were collected using the blunt heat sealed end of Pasteur pipette and transferred to a drop of 0.25% trypsin 0.02% EDTA under mineral oil.These were then incubated for 3-4 min at 37ºC, 5% Co2 and 95% air. After that the cells were separated using very fine Pasteur pipette filled with DMEM medium and washing the cells several times with fresh medium. The isolated cells were transferred to 96 well dishes containing DMEM supplemented with 10% FBS and 10% newborn calf serum (NCS), 0.1 mM β mercaptoethanol and 1000 U/ml LIF. Cultures were incubated for two weeks at 37ºC in water saturated atmosphere and 5% Co2. Medium was renewed after two or three days. Each well was examined every day up to 14 days. The colonies having typical EscMorphology were subcultured. The sub culturing method was similar to the previous procedure.
Identification of ES cells
Histochemical staining for alkaline phosphatase was carried out as described by Donovan et al, (1986). Materials for this technique were purchased from Sigma company (Sigma, St Louis, USA). Briefly, the cytospine cell population was fixed with acetone formaldehyde solution (1 min), then washed with deionised water and incubated with 1mg/ml Fast Red TR salt and 40 µl/ml Naphtol As-Mx phosphate at pH of 8.4 for 5 min. After washing and counter staining using hematoxylin solution (2 min), the samples were examined under light microscope.Results
The summary of embryo development and formation of ICM and ES colonies are presented in Table I. 78% of embryos were hatched following incubation in DMEM for 12-14 h (Figure 1). After hatching, there were two morphologically distinct cell types, ICM colonies and trophectoderm. Later, cells
were attached to the bottom of plate as a monolayer. The ICM cell colonies were small cells with a large nucleus and minimal cytoplasm (Figure 2). These cells were tightly packed and before trypsinizing were
were tightly packed and before trypsinizing were difficult to recognize individual cells. The morphology of ICM colonies in the first, second and third subcultures were similar (Figure 3). However, they formed monolayer on the plate following forth subculture (Figure 4). 89% of the hatched embryos formed ICM colonies; however, only 10% of ICM colonies could produce ESc. Red staining of the cells after alkaline phosphatase histochemistry, indicated a positive enzyme reaction, which demonstrated in Figure 5.
Discussion
The main purpose of this study was to establish the isolation and production of NMRI mouse ES cells. Our results showed that the ICM colonies that were obtained from hatched NMRI mouse blastocyst could produce ES cells. After some sub culturing, the morphology of those cells were similar to the other ES cell lines, which have been obtained from another strain of mammals such as human (Bongso et al.,1994; Thomson et al., 1995; Doetschman et al., 1988). The alkaline phosphatase reaction showed that these cells became undifferentiated after some subculturing.
Our result also demonstrated that LIF in the absence of feeder layer is efficient in preventing the differentiation of ES cells. In contrast, Anderson (1992) showed an unsuccessful experiments for usage of murine and human LIF to assist isolation of ESc. Therefore, it appears that the action of LIF on ICM cells may be species as well as strain dependent.
In conclusion, the results showed that a highly pluripotent ESc can be derived from blastocyst of NMRI mice. It was also demonstrated that these cells could be differentiated into GM-CSF hematopoietic colonies (Movassagh Pour et al., 2003). Thus, this technique could be effective to produce ESc which may be used in the future reproductive studies.
Acknowledgement
The authors would like to thank Mr M. Solaymani for technical assistance. This study was funded by Iran Blood Transfusion Organization.
Type of Study: Original Article |
References
1. Anderson GB. (1992) Isolation and use of embryonic stem cells from livestock species. Anim Biotechnol 3: 165-175. [DOI:10.1080/10495399209525769]
2. Bongso A, Fong CY, Ng SC, Ratnam S. (1994) Isolation and culture of inner cell mass cells from human blastocysts. Hum Reprod 9: 2119-2117. [DOI:10.1093/oxfordjournals.humrep.a138401] [PMID]
3. Cole RJ, Edwards RC, Poul J. (1965) Cytodifferentiation in cell colonies and cell strains derived from cleaving ova and blastocysts of the rabbit. J Exp Cell Res 501-504. [DOI:10.1016/0014-4827(65)90201-6]
4. Doetschman T, Williams P, Maeda N. (1988) Establishment of hamster blastocyst derived embryonic stem cells. Dev Biol 127: 224-227. [DOI:10.1016/0012-1606(88)90204-7]
5. Donovan PJ, Stott D, Cairns LA, Heasman J, Wylie CC. (1986) Migratory and postmigratory mouse primordial germ cells behave differently in culture. Cell 44: 831-838. [DOI:10.1016/0092-8674(86)90005-X]
6. Evans MJ, Kaufman M. (1981) Establishment in culture of pluripotential cells from mouse embryos. Nature 242: 154-156. [DOI:10.1038/292154a0] [PMID]
7. Martin GR. (1981) Isolation of a pluripotent cell line from early mouse embryos cultured in medium contained with teratocarcinoma stem cells. Proc Nat Sci 78: 7634-7638. [DOI:10.1073/pnas.78.12.7634] [PMID] [PMCID]
8. Movassagh Pour A, Salehnia M, Pourfatollah M, Moazzeni SM. (2003) CFU-GM like colonies derived from embryonic stem cells cultured on the bone marrow stromal cells. Iranian Biomedical J (In press).
9. Odorico JS, Kaufman DS, Thomson JA. (2001) Multilineage differentiation from human embryonic stem cell lines. Stem Cells 19: 193-204. [DOI:10.1634/stemcells.19-3-193] [PMID]
10. Pera MF, Reubinoff BE, Truonson A. (2000) Human embryonic stem cells. Cell Science 113: 5-10.
11. Rathjeh PD, Lake J, Whyatt, Bettess MD, Rathjeh J. (1998) Properties and uses of embryonic stem cells: prospects for application to human biology and gene therapy. Reprod Fertil Dev 10: 31-47. [DOI:10.1071/R98041] [PMID]
12. Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. (2000) Embryonic stem cell lines from human blastocyst: somatic differentiation in vitro. Nature Biotech. 18: 399-404. [DOI:10.1038/74447] [PMID]
13. Smith AG, Hooper L. (1987) Buffalo liver cell produce a diffusible activity which inhibits the differentiation of murine embryonal carcinoma and embryonic stem cells. Dev Biol 121: 1-9. [DOI:10.1016/0012-1606(87)90132-1]
14. Smith AG, Heath JK, Donaldsmon DD, Wong GG, Moreau J, Stahl M, Rogers D. (1988) Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 336: 688-690. [DOI:10.1038/336688a0] [PMID]
15. Smith AG. (1998) Cell therapy: In search of pluripotency. Current Biol 8; 802-804. [DOI:10.1016/S0960-9822(07)00504-0]
16. Smith AG. (1992) Mouse embryo stem cells: their identification, propagation and manipulation. Seminar in Cell Biol 3: 385-399. [DOI:10.1016/1043-4682(92)90010-S]
17. Thomson JA, Marshal VS. (1998) Primate embryonic stem cells. Curr Topics Dev Biol 38: 133-165. [DOI:10.1016/S0070-2153(08)60246-X]
18. Thomson JA, Kalishman J, Golos TS, Durning M, Harris CP, Becker RA, Hearn JP. (1995) Isolation of a primate embryonic stem cell line. Proc Natl Acad Sci USA 92: 7844-48. [DOI:10.1073/pnas.92.17.7844] [PMID] [PMCID]
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
