
International Journal of
Reproductive Biomedicine
Tue, Aug 4, 2026
[Archive]
Volume 22, Issue 6 (June 2024)
IJRM 2024, 22(6): 495-506 |
Back to browse issues page



Ethics code: IR.SKU.REC.1402.020
![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Ahmadi K, Reiisi S, Habibi Z. Comparison of the gene expression profiles of endometrial and trophoblastic cells in women with recurrent miscarriage: A bioinformatics approach. IJRM 2024; 22 (6) :495-506
URL: http://ijrm.ir/article-1-3136-en.html
URL: http://ijrm.ir/article-1-3136-en.html



1- Department of Computer Sciences, Faculty of Mathematical Sciences, Shahrekord University, Shahrekord, Iran.
2- Department of Genetics, Faculty of Basic Sciences, Shahrekord University, Shahrekord, Iran. ,s.reiisi@yahoo.com
3- Department of Women and Family Affairs, Chaharmahal and Bakhtiari Governorate, Shahrekord, Iran.
2- Department of Genetics, Faculty of Basic Sciences, Shahrekord University, Shahrekord, Iran. ,
3- Department of Women and Family Affairs, Chaharmahal and Bakhtiari Governorate, Shahrekord, Iran.
Full-Text [PDF 1452 kb]
(1156 Downloads)
| Abstract (HTML) (1516 Views)
Full-Text: (280 Views)
- Introduction
Recurrent miscarriage (RM) is characterized by terminating all identified pregnancies within the uterus (1, 2). The treatment of RM has become a challenging issue in reproductive medicine due to the limited understanding of its pathogenesis. Numerous advances in genetics, immunology, and cell biology have revealed the significant involvement of genes and noncoding RNAs in the onset and progression of RM.
The regulation of cellular processes, including cell cycle, apoptosis, and epithelial-mesenchymal transition, by various transcriptomic networks can impact placental trophoblast growth, migration, and invasion (3). Hence, aberrant expression of various transcriptomic factors may contribute to the onset and progression of recurrent spontaneous miscarriage.
During the primary phases of a typical pregnancy, the proper execution of various functions by the fetal trophoblast significantly impacts the fetus viability (4). Following the successful implantation of the embryo, a process of cellular differentiation arises, whereby the extravillous trophoblast infiltrate the maternal uterine tissue, establishing a fixed attachment of the placenta to the uterus. This event is accompanied by restructuring of the maternal vasculature, which facilitates the provision of vital nutrients to the developing fetus. Any mistake made during this process can result in pathological pregnancies, which may include recurrent spontaneous miscarriages (5, 6). The necessity of cell signaling appears to be evident in facilitating communication between trophoblast and endometrium. Alterations in the concentration of intracellular signaling molecules resulting from genetic modifications and correlated transcripts may give rise to gestational disorders, including RM, pre-eclampsia, and fetal growth restriction (7).
Transcriptome production marks the initial stage of any cell function or structure alteration. Variations in cellular function are attributed to alterations in gene expression. Hence, gene expression analysis can offer a valuable understanding of the disease pathogenesis (8). Transcriptomics investigations have the potential to provide numerous genes as molecular markers (9).
Identifying biomarkers and understanding the complicated process of embryo implantation may facilitate the classification of distinct phases of endometrial acceptance (pre- and post-implantation) and the formulation of tailored pharmacological interventions. Furthermore, the detection of genes that exhibit differential expression genes (DEG) holds significant value in the identification of enrichment pathways, which can aid in the interpretation of molecular processes that have been altered in cases of abortion and the comprehension of anomalous mechanisms of the disease (10).
The present study aimed to examine genes exhibiting alterations in expression within 2 distinct cell groups (endometrium and trophoblast) using analyzing expression data obtained from the gene expression omnibus database. Subsequently, a comparative analysis is conducted between the genetic profiles of both groups, focusing on identifying common and distinct genes.
The following step involves the identification of the biological pathways that are associated with each group of genes.
The regulation of cellular processes, including cell cycle, apoptosis, and epithelial-mesenchymal transition, by various transcriptomic networks can impact placental trophoblast growth, migration, and invasion (3). Hence, aberrant expression of various transcriptomic factors may contribute to the onset and progression of recurrent spontaneous miscarriage.
During the primary phases of a typical pregnancy, the proper execution of various functions by the fetal trophoblast significantly impacts the fetus viability (4). Following the successful implantation of the embryo, a process of cellular differentiation arises, whereby the extravillous trophoblast infiltrate the maternal uterine tissue, establishing a fixed attachment of the placenta to the uterus. This event is accompanied by restructuring of the maternal vasculature, which facilitates the provision of vital nutrients to the developing fetus. Any mistake made during this process can result in pathological pregnancies, which may include recurrent spontaneous miscarriages (5, 6). The necessity of cell signaling appears to be evident in facilitating communication between trophoblast and endometrium. Alterations in the concentration of intracellular signaling molecules resulting from genetic modifications and correlated transcripts may give rise to gestational disorders, including RM, pre-eclampsia, and fetal growth restriction (7).
Transcriptome production marks the initial stage of any cell function or structure alteration. Variations in cellular function are attributed to alterations in gene expression. Hence, gene expression analysis can offer a valuable understanding of the disease pathogenesis (8). Transcriptomics investigations have the potential to provide numerous genes as molecular markers (9).
Identifying biomarkers and understanding the complicated process of embryo implantation may facilitate the classification of distinct phases of endometrial acceptance (pre- and post-implantation) and the formulation of tailored pharmacological interventions. Furthermore, the detection of genes that exhibit differential expression genes (DEG) holds significant value in the identification of enrichment pathways, which can aid in the interpretation of molecular processes that have been altered in cases of abortion and the comprehension of anomalous mechanisms of the disease (10).
The present study aimed to examine genes exhibiting alterations in expression within 2 distinct cell groups (endometrium and trophoblast) using analyzing expression data obtained from the gene expression omnibus database. Subsequently, a comparative analysis is conducted between the genetic profiles of both groups, focusing on identifying common and distinct genes.
The following step involves the identification of the biological pathways that are associated with each group of genes.
- Materials and Methods
2.1. Data collection
This bioinformatics study involved retrieving data relating to endometrium and trophoblast from the gene expression omnibus database (http://www.ncbi.nlm.nih.gov/geo/) after initial inquiries. This study investigated 2 different datasets for the problem of recurrent abortion and found an overlap between these 2 datasets. In this approach, better treatment or diagnostic markers can be provided. The dataset concerning the endometrium, identified by the accession code GSE165004, comprises 24 subjects with normal fertility and 24 with RMs. The study of GSE165004 was directed to understand the transcriptomic profile of mid-secretory phase endometria of patients with recurrent pregnancy losses and unexplained infertility by comparing with the endometria of healthy fertile women (controls) by weighted gene co-expression network analysis. However, in our study, only data related to recurrent abortions and healthy women were examined with a different method.
Additionally, the trophoblast data, accessible through the GSE76862 accession code, consists of 3 healthy controls and 3 subjects with RMs. The study included information relating to both asymptomatic women and women with a history of RM, determined by a specialist. The inclusion criteria for the study contained individuals who lacked any abnormalities of uterine and fallopian tubes or obstructions in various uterine segments; moreover, these individuals exhibited a normal karyotype. This study aimed to find the common pathway between 2 important parts in fertility, that is, endometrial and trophoblast cells; however, with the inclusion criteria, there was only one data in the database for each case. Therefore, it was impossible to add more data. Other datasets had different samples or genetic markers.
This bioinformatics study involved retrieving data relating to endometrium and trophoblast from the gene expression omnibus database (http://www.ncbi.nlm.nih.gov/geo/) after initial inquiries. This study investigated 2 different datasets for the problem of recurrent abortion and found an overlap between these 2 datasets. In this approach, better treatment or diagnostic markers can be provided. The dataset concerning the endometrium, identified by the accession code GSE165004, comprises 24 subjects with normal fertility and 24 with RMs. The study of GSE165004 was directed to understand the transcriptomic profile of mid-secretory phase endometria of patients with recurrent pregnancy losses and unexplained infertility by comparing with the endometria of healthy fertile women (controls) by weighted gene co-expression network analysis. However, in our study, only data related to recurrent abortions and healthy women were examined with a different method.
Additionally, the trophoblast data, accessible through the GSE76862 accession code, consists of 3 healthy controls and 3 subjects with RMs. The study included information relating to both asymptomatic women and women with a history of RM, determined by a specialist. The inclusion criteria for the study contained individuals who lacked any abnormalities of uterine and fallopian tubes or obstructions in various uterine segments; moreover, these individuals exhibited a normal karyotype. This study aimed to find the common pathway between 2 important parts in fertility, that is, endometrial and trophoblast cells; however, with the inclusion criteria, there was only one data in the database for each case. Therefore, it was impossible to add more data. Other datasets had different samples or genetic markers.
2.2. DEG analysis in the endometrium and trophoblast
The limma package of R software was employed for analyzing microarray and RNA-seq data. The RMA method for microarray data and counts per million for RNA-seq data were used for normalization and filtering. Initially, an analysis was conducted on the endometrium sample data. Subsequently, the dataset concerning trophoblast was also analyzed following the identification of DEGs. The statistical significance threshold for screening both datasets was determined as adj-p-value < 0.05 and |log2FC| > 0.5.
Subsequently, the EnhancedVolcano package in the R programming language generated volcano plots representing DEGs. Following the identification of DEGs in each group using a Venn diagram, the overlapped genes between the 2 groups were subsequently determined.
Subsequently, the EnhancedVolcano package in the R programming language generated volcano plots representing DEGs. Following the identification of DEGs in each group using a Venn diagram, the overlapped genes between the 2 groups were subsequently determined.
2.3. Enrichment analysis of target genes
The gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for both endometrial and trophoblast were determined using the clusterProfiler package and KEGG Orthology-Based Annotation System (KOBAS) online database for common and distinct genes. The cluster profile software package employs the R programming language to analyze GO, which comprises biological processes, molecular functions, and cellular components. The KOBAS database employs machine learning to rank biologically significant pathways effectively. The KOBAS server employs a total of 10 databases for analysis, consisting of 5 pathway databases (KEGG Pathway, PID, BioCyc, Reactome, and Panther) and 5 human databases (OMIM, KEGG Disease, FunDO, GAD, and NHGRI GWAS). To investigate this analysis, both common and distinct genes were uploaded into the database, and enrichment analyses were subsequently conducted.
2.4. Protein-protein interaction (PPI) network and analysis of key genes
PPI concerns the intermolecular interaction between proteins within biological processes. The STRING database, which stands for the retrieval of interacting genes and is accessible via https://string-db.org/, is a web-based server utilized to assess PPI network data. To achieve this objective, the overlapped and distinct genes among the groups were identified, then the network was constructed by utilizing the STRING platform. Subsequently, the corresponding network was generated by Cytoscape software version 3.9.1. The CutoHabba plugin was used within the Cytoscape software platform to determine the hub genes in the network exhibiting the highest score.
2.5. Ethical considerations
This study was ethically approved by the Ethical Committee of Shahrekord University, Shahrekord, Iran (Code: IR.SKU.REC.1402.020).
- Results
3.1. Analysis of differential gene expression data in the endometrium and trophoblast
Before conducting a differential analysis of gene expression, a quality assessment and normalization of the expression datasets (GSE165004 and GSE76862) were accomplished. To assess data quality, zero values in each sample were surveyed, and subsequently, data relating to zero-valued items were eliminated through filtration. The normalization of data was performed utilizing the techniques provided by the limma package.
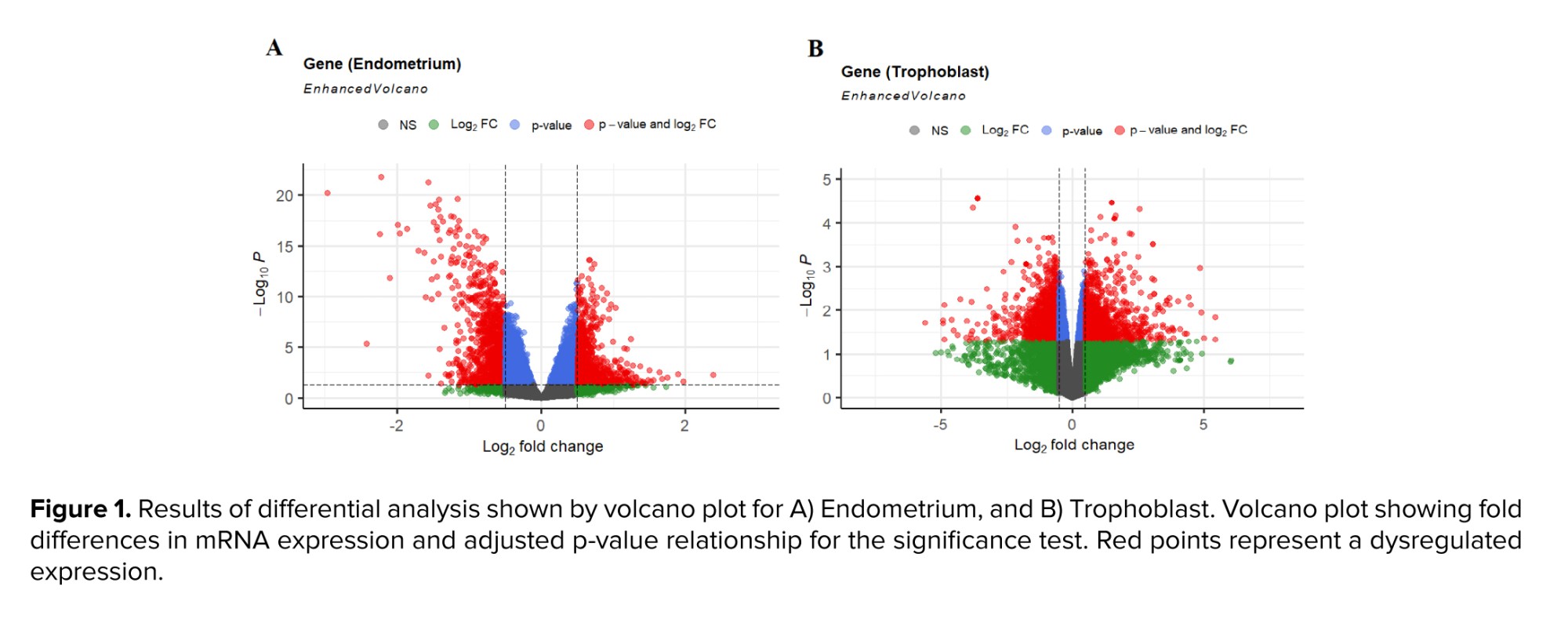
Upon analysis of the corrected expression data and utilizing the established significance threshold of p < 0.05 and |log2FC| > 0.5, 1701 genes were identified as having undergone alterations in expression within the trophoblast dataset. Specifically, 751 genes exhibited increased expression, while 950 genes displayed decreased expression. The study analyzed endometrium data, revealing a total of 1041 genes with differential expressions. Among these genes 480 exhibited upregulation, while 561 exhibited downregulation.
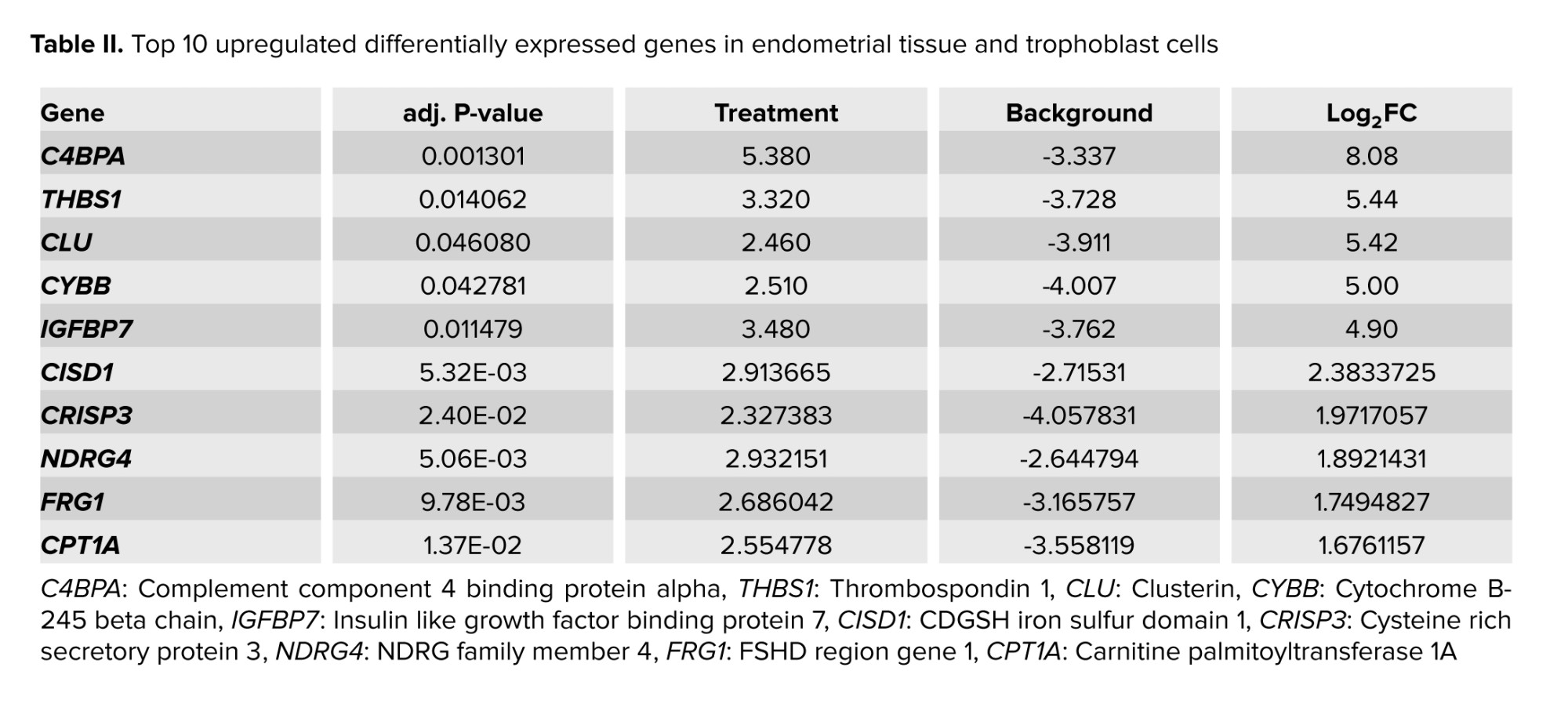
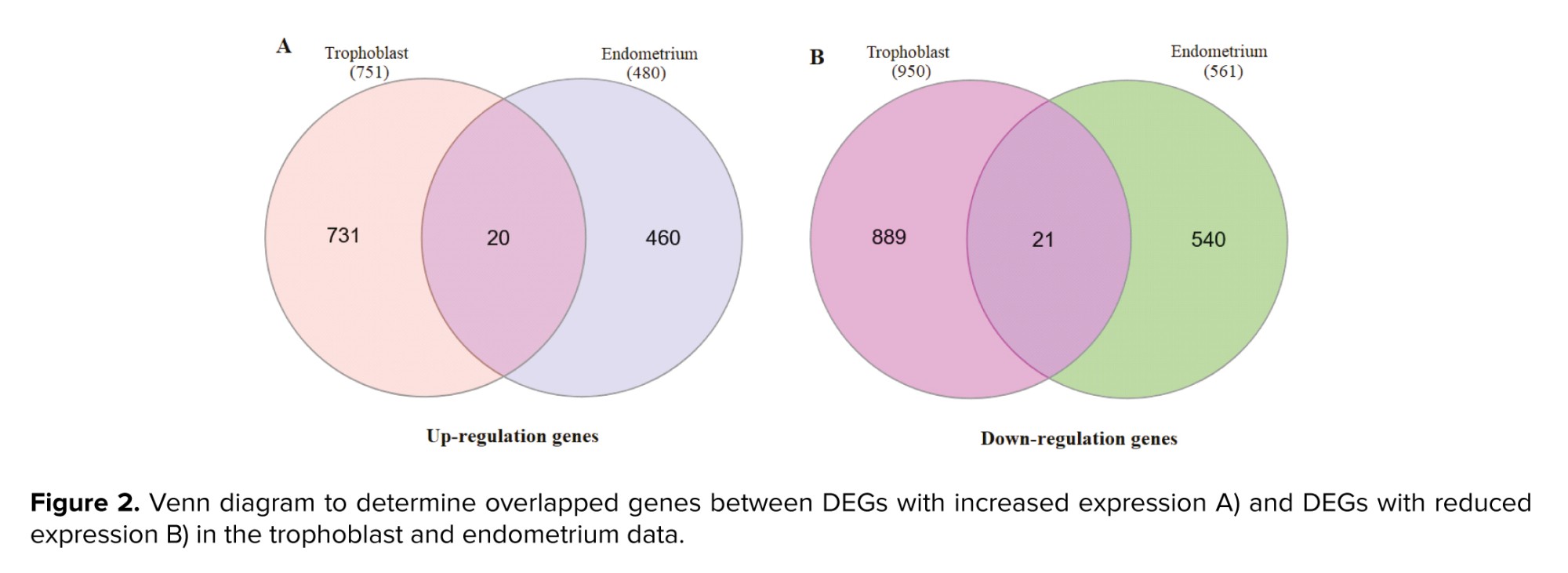
Tables I, and II display the 10 genes with the highest score in up- or downregulation across both datasets. Furthermore, figure 1 shows volcano plots for the analyzed data, with the red data points indicating instances of differential expression. The Venn diagram determined the overlapped genes between 2 distinct groups. Figure 2 reveals that 21 common genes were observed between trophoblast and endometrium DEGs exhibiting decreased expression, while 20 common genes were identified between DEGs exhibiting increased expression.
3.2. Gene enrichment analysis
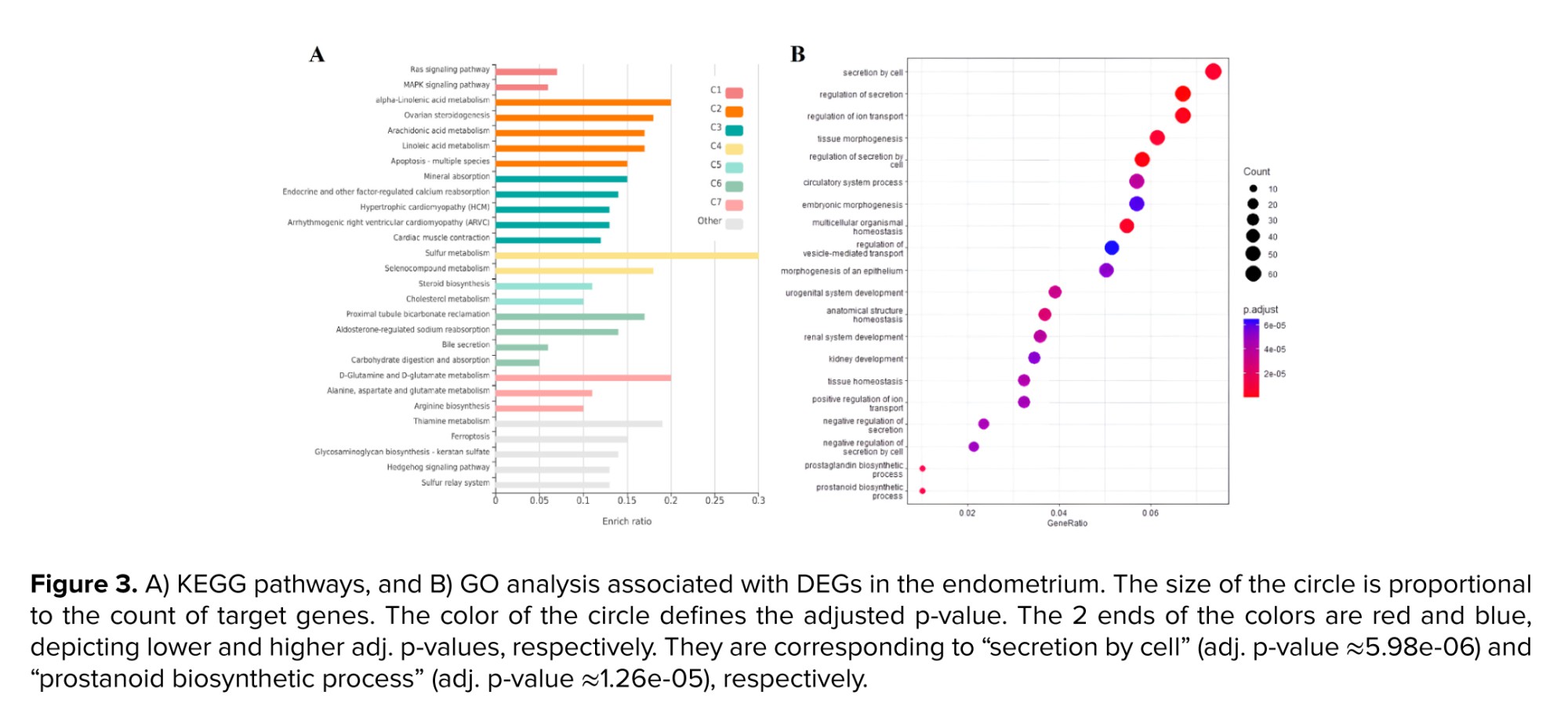
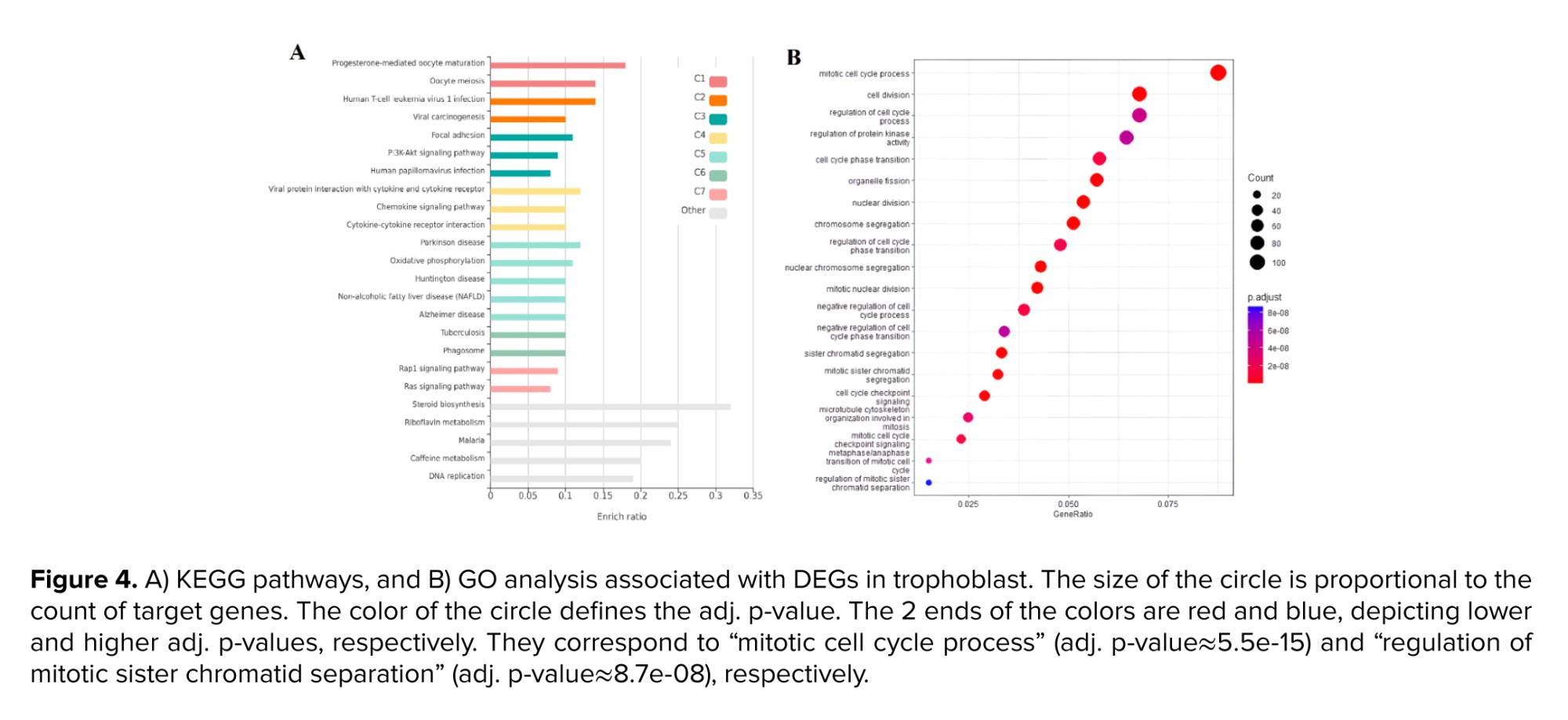
To conduct a more comprehensive examination of overlapped genes between trophoblast and endometrium data, as well as unique genes within each group, GO analysis and analysis of signaling pathways were carried out utilizing the KEGG database. The survey results revealed 20 pathways that obtained the highest score. As per the findings presented in figure 3A, the distinct genes associated with the endometrium, the KEGG database has identified various signaling pathways that include the synthesis and metabolism of compounds such as sulfur, thiamine, cholesterol, linoleic acid, arachidonic acid, and solenoid compounds. Furthermore, apoptosis and cell cycle pathways were most significantly recognized among the pathways. According to the GO analysis conducted on these genes, significant findings were observed in relation to the secretion of cell factors, ion transport, tissue morphogenesis, and embryonic and epithelial cells (Figure 3B). The KEGG pathways analysis revealed significant enrichment of viral infection pathways, including papillomavirus and virus interaction with various receptors, phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway, cell division, central junctions, and chemokine-dependent signaling in trophoblast (Figure 4A).
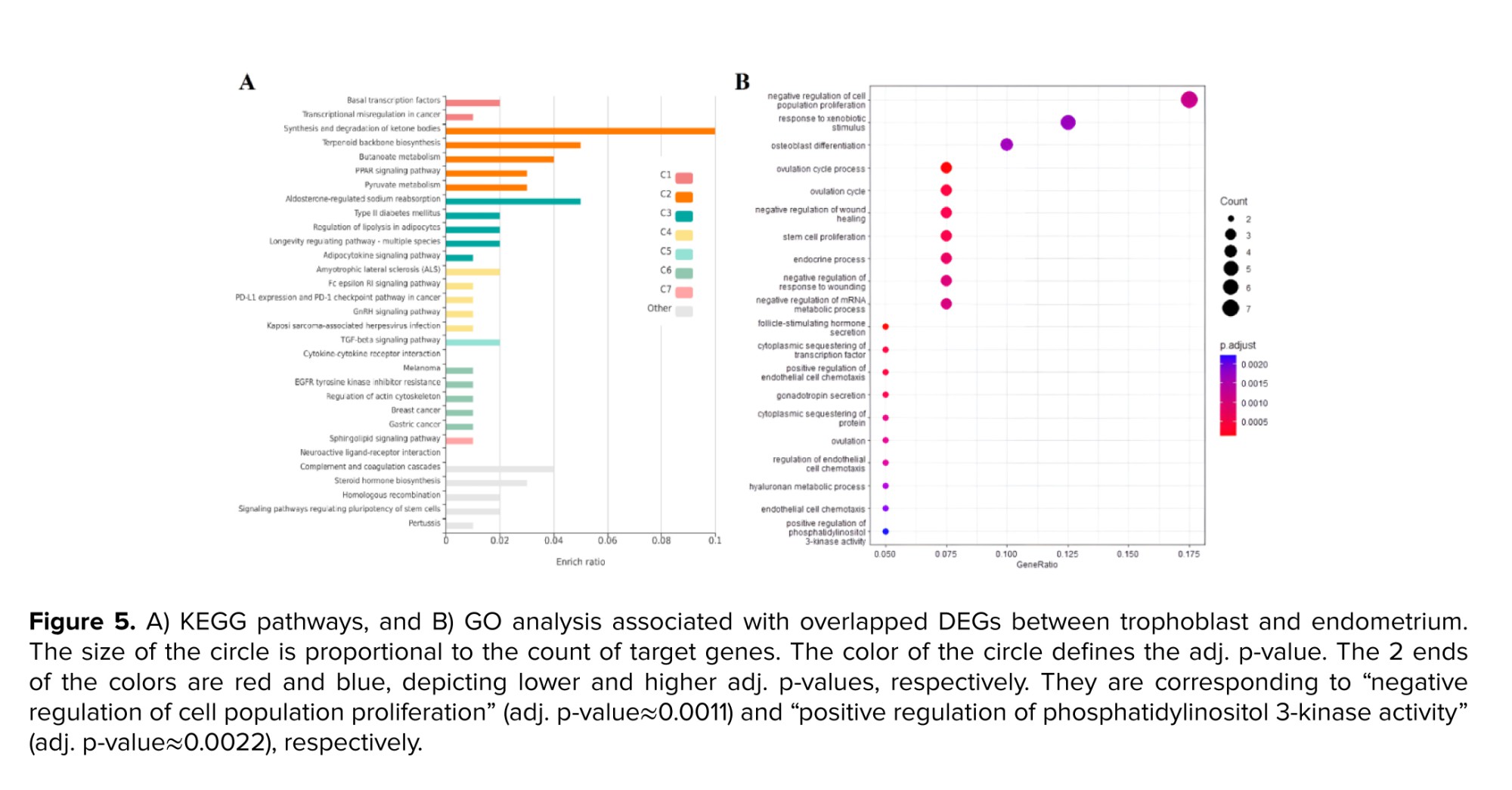
The GO analysis in the trophoblast cells revealed a significant enrichment of various items related to cell division, cell cycle, and associated processes, including chromosomal division and separation, as well as the transfer of different phases of the cell cycle (Figure 4B). Upon analyzing common genes, it was observed that a significant number of genes contributed to the biosynthesis and catabolism of ketone bodies, as indicated by the KEGG pathway analysis. Several pathways were identified, including the biosynthesis of terpenoids, pyruvate metabolism, and a cascade related to coagulation and complement (Figure 5A). The study conducted a GO analysis on common genes, which yielded significant findings such as the inhibition of cell proliferation, response to xenobiotic stimuli, and negative regulation of wound healing and stem cell proliferation (Figure 5B).
3.3. PPI network
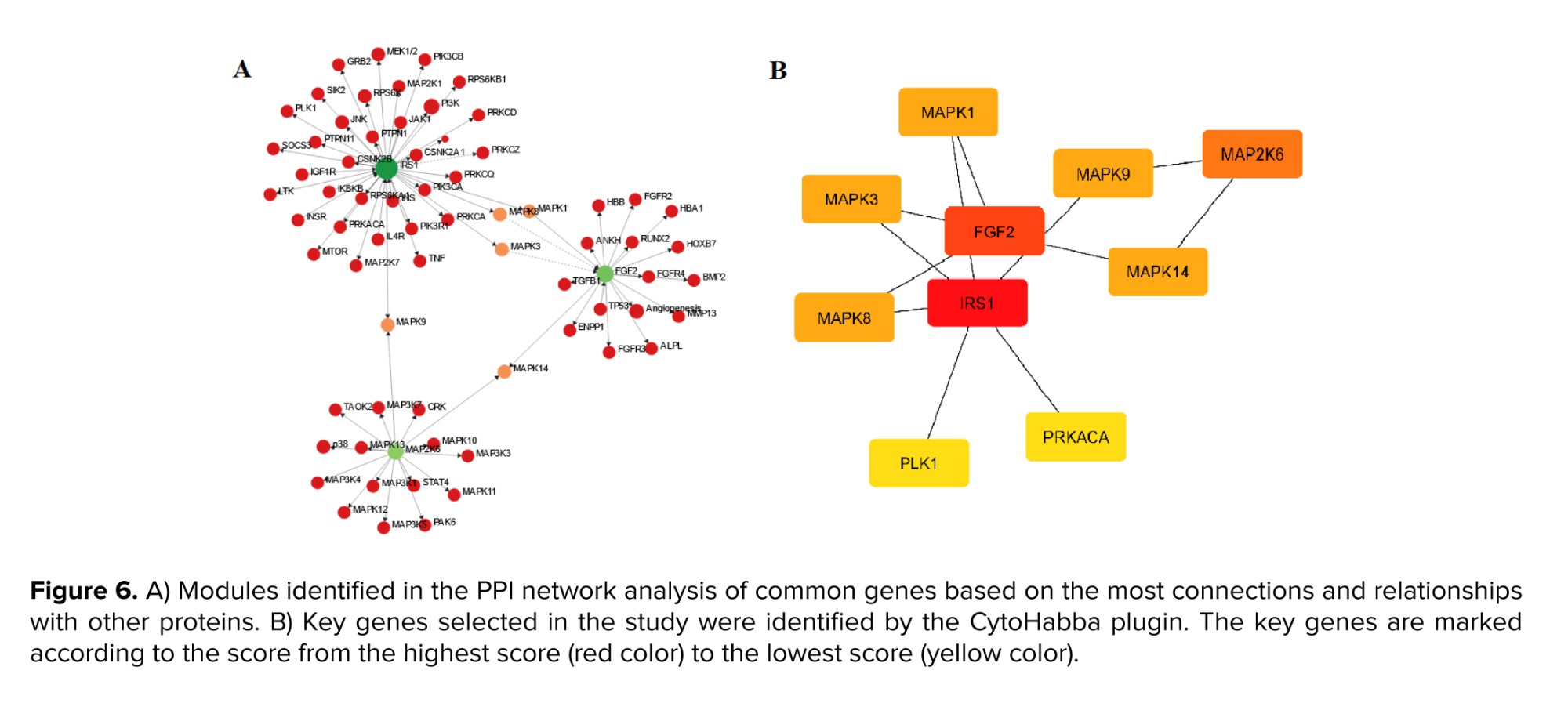
The interaction data relating to overlapped genes, and the PPI network were acquired using the STRING database. A network was constructed to represent shared genes, consisting of 74 nodes and 130 edges with a degree of 0.8. The network underwent analysis using Cytoscape software, extracting a key network with the highest score from the STRING network (Figure 6A). The CytoHabba plugin within the Cytoscape software was utilized to identify hub genes. Note that the plugin employs various topological techniques, and in the current study, the degree approach was utilized to identify hub genes. The method used is measured precisely in its identification of crucial proteins, as it relies on the highest degree of connections within the network and subsequently assigns them the highest score (Figure 6B). The key genes identified as significant through analysis in the Cytoscape software were insulin receptor substrate 1 (IRS1), fibroblast growth factor 2 (FGF2), mitogen-activated protein kinase 6 (MAPK6), MAPK1, MAPK3, MAPK8, MAPK9, polo like kinase 1 (PLK1), protein kinase CAMP-activated catalytic subunit alpha (PRKACA), and protein kinase C alpha (PRKCA).

4. Discussion
The present study involved selecting and examining 2 distinct sets of samples. The initial cohort comprised endometrium samples, while the subsequent cohort consisted of trophoblasts that were extracted from women who had experienced RMs and healthy controls. Upon conducting a comprehensive analysis of the data, a comparative analysis was performed between the 2 distinct sample types, identifying 41 common genes between the endometrium and trophoblast.
The problem of RMs remains a significant reproductive concern worldwide and a prevalent complication associated with pregnancy.
To understand the abnormality in the fertility and reproductive system leading to fetal loss, it is vital to examine the mechanisms and factors implicated in the ovarian follicle production process and the implantation of the fetus in the maternal uterus, as well as the disorders of these pathways. The processes are greatly influenced by genetics. While the diagnosis of RMs may be challenging, it can be beneficial for both the medical practitioner and the patient, as it enhances the chance of a successful subsequent pregnancy. Thus, identifying effective factors in this regard may be highly advantageous.
Upon studying the pathways implicated in the functioning of overlapped genes, a considerable proportion of genes exhibiting alterations in expression are subject to regulation within the cell cycle pathway. Disruptions in this pathway are associated with the activation of cellular proliferation and the inhibition of apoptosis, as previously reported (11). Moreover, it has been revealed that cell cycle regulation plays a crucial role in the differentiation process of uterine stromal cells during implantation. The genes responsible for regulating the cell cycle exhibit dysregulation during the secretory phase of the endometrium. Hence, regulating gene expression relating to this process through hormone intervention, such as progesterone or steroids, has been observed to be effective in enhancing the implantation rate (12). Also, one of the findings elucidated in this investigation relates to the function of apoptosis mediated by the involved genes. Comparative analyses between samples with miscarriages and healthy samples have revealed a significant increase in the level of apoptosis in the former group. Subsequent investigations have suggested that increased apoptosis may be one of the factors contributing to RMs (13). Apoptosis has been verified to exist in trophoblast during the initial stages of pregnancy. The processes of implantation, blastocyst growth, endometrial regeneration, and placental structure reconstruction, among others, are intricately linked to apoptosis (14).
The balance between cellular proliferation in placental villi and endometrium and the level of apoptosis is crucial in pregnancy physiology. However, in pathological conditions such as miscarriage, this process is disrupted and influenced, leading to an imbalance. The present study displays the PI3K-Akt signaling pathway, which regulates cellular growth and apoptosis. Dysregulation of this pathway increases proliferation or apoptosis within the endometrium (15). Typically, the PI3K-Akt signaling pathway is downregulated in initiating embryo acceptance by the endometrium. During the proliferative phase, the PI3K-Akt signaling pathway expression is diminished in women with a thin endometrium (16).
Thus, the aforementioned approach may also prove effective in abortion. An additional pathway identified is the metabolic pathway of ketone bodies. Ketone bodies are a class of organic compounds characterized by a carbonyl group in the middle of the carbon chain. They are synthesized in the hepatic tissue through the process of ketogenesis. Ketone bodies are a significant substitute fuel for peripheral tissues, particularly in the absence of glucose. Various factors, including glucagon trigger the induction of lipolysis and ketogenesis, while insulin suppresses these processes (17). Research has indicated that pregnant women are at a higher risk of experiencing ketosis than others (18). Numerous investigations, comprising animal model experiments, have demonstrated that environments characterized by elevated ketone levels may result in unfavorable outcomes for both the mother and the fetus (19). The study found that beta-hydroxy-butyric acid exposure in mouse embryos resulted in a range of abnormalities, including growth retardation and neural tube defects. The severity of these abnormalities was found to increase with higher exposure doses (20).
The pathway associated with the activation of the complement system was highly significant among the pathways linked to shared genes. The complement system exhibits activity throughout the menstrual cycle, with heightened expression observed during the secretory phase. The molecules of the complement system impact the attachment between the embryo and the endometrium at the location of implantation. The complement protein C1q has a significant function in the implantation process within the endometrium and is also implicated in the pathogenesis of pre-eclampsia, as evidenced by previous research (21). It have demonstrated that the coagulation and complement cascade pathway is significant in the endometrium during pregnancy (22). Apart from preserving the endometrium through the preservation of epithelial integrity, the presence of a functional complement system also raises worries regarding the possibility of the fetus being perceived as a foreign object. Thus, it is crucial to establish balance in this case (23).
Furthermore, hub genes were identified in the pathways discovered during the study. IRS1 and FGF2 were identified as common genes with the highest score. The IRS1 gene encodes for the insulin receptor and initiates the insulin-dependent signaling cascade. Insulin is a crucial metabolic hormone in maintaining energy homeostasis within the human body (24). The insulin-dependent signaling pathway is a crucial factor in the development of the fetus. The insulin hormone, acting as a growth factor, can potentially enhance cell proliferation and inhibit apoptosis via the PI3K-Akt pathways in endometrial cancer (25). The invasion behavior of endometrial cancer is associated with the activation of the insulin receptor IRS1. Over the initial 20 wk of development, there is a steady increase in fetal growth hormone levels, which significantly impacts insulin metabolism (26). Hence, the balance of energy can impact the embryo implantation process and subsequent growth. The current study demonstrates the reduction in the insulin receptor gene IRS1 expression among females who experienced miscarriages. The FGF2 gene is responsible for encoding the fibroblast growth factor- biomolecules that induce cellular growth, proliferation, and homeostatic regulation. These factors are crucial in regulating proliferation, differentiation, angiogenesis, tissue regeneration, and embryo development (27).
The expression of FGF2 is observed to be elevated in the endometrium during the gestational period. The literature indicates that FGF2 has demonstrated the ability to induce trophoblast migration, adhesion, and embryogenesis across multiple species (28). In addition, it has been established that FGF2, in conjunction with vascular endothelial growth factor, plays a crucial role in regulating the angiogenesis process during placental development (29). The observed reduction in FGF2 expression in samples associated with RMs highlights this factor's significance in maintaining fetal viability throughout gestation.
5. Conclusion
The current study aimed to employ bioinformatics methodologies and tools to identify significant genes and their associated pathways in endometrium and trophoblasts. Furthermore, a comparative analysis was conducted to identify the common genes between the 2 groups by examining the genes exhibiting alterations in expression across both tissues. The study's findings indicate that the genes analyzed in both endometrial and trophoblastic samples are significant in women with RMs compared to those with normal pregnancies. Conversely, the co-occurrence of common genes in the aforementioned groups simultaneously underscores the significance of accuracy in both cell types.
Data availability
The data generated during and/or analyses during the current study are available from the corresponding author upon reasonable request.
Author contributions
K. Ahmadi performed most of the bioinformatics data analysis. S. Reiisi coordinated the study, participated in intellectual discussions of the data, wrote and revised the manuscript. Z. Habibi participated in intellectual discussions of the data and helped in reviewing the manuscript. All authors approved the final manuscript and take responsibility for the integrity of the data.
Acknowledgements
We acknowledged all the individuals who helped do this study, particularly the Research Vice-Chancellor for Research and Innovation of Shahrekord University, Shehrekord, Iran. Artificial intelligence was used to improve the language and readability of the paper.
Conflict of Interest
The authors declare that they have no conflict of interest.
Before conducting a differential analysis of gene expression, a quality assessment and normalization of the expression datasets (GSE165004 and GSE76862) were accomplished. To assess data quality, zero values in each sample were surveyed, and subsequently, data relating to zero-valued items were eliminated through filtration. The normalization of data was performed utilizing the techniques provided by the limma package.
Upon analysis of the corrected expression data and utilizing the established significance threshold of p < 0.05 and |log2FC| > 0.5, 1701 genes were identified as having undergone alterations in expression within the trophoblast dataset. Specifically, 751 genes exhibited increased expression, while 950 genes displayed decreased expression. The study analyzed endometrium data, revealing a total of 1041 genes with differential expressions. Among these genes 480 exhibited upregulation, while 561 exhibited downregulation.
Tables I, and II display the 10 genes with the highest score in up- or downregulation across both datasets. Furthermore, figure 1 shows volcano plots for the analyzed data, with the red data points indicating instances of differential expression. The Venn diagram determined the overlapped genes between 2 distinct groups. Figure 2 reveals that 21 common genes were observed between trophoblast and endometrium DEGs exhibiting decreased expression, while 20 common genes were identified between DEGs exhibiting increased expression.
3.2. Gene enrichment analysis
To conduct a more comprehensive examination of overlapped genes between trophoblast and endometrium data, as well as unique genes within each group, GO analysis and analysis of signaling pathways were carried out utilizing the KEGG database. The survey results revealed 20 pathways that obtained the highest score. As per the findings presented in figure 3A, the distinct genes associated with the endometrium, the KEGG database has identified various signaling pathways that include the synthesis and metabolism of compounds such as sulfur, thiamine, cholesterol, linoleic acid, arachidonic acid, and solenoid compounds. Furthermore, apoptosis and cell cycle pathways were most significantly recognized among the pathways. According to the GO analysis conducted on these genes, significant findings were observed in relation to the secretion of cell factors, ion transport, tissue morphogenesis, and embryonic and epithelial cells (Figure 3B). The KEGG pathways analysis revealed significant enrichment of viral infection pathways, including papillomavirus and virus interaction with various receptors, phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway, cell division, central junctions, and chemokine-dependent signaling in trophoblast (Figure 4A).
The GO analysis in the trophoblast cells revealed a significant enrichment of various items related to cell division, cell cycle, and associated processes, including chromosomal division and separation, as well as the transfer of different phases of the cell cycle (Figure 4B). Upon analyzing common genes, it was observed that a significant number of genes contributed to the biosynthesis and catabolism of ketone bodies, as indicated by the KEGG pathway analysis. Several pathways were identified, including the biosynthesis of terpenoids, pyruvate metabolism, and a cascade related to coagulation and complement (Figure 5A). The study conducted a GO analysis on common genes, which yielded significant findings such as the inhibition of cell proliferation, response to xenobiotic stimuli, and negative regulation of wound healing and stem cell proliferation (Figure 5B).
3.3. PPI network
The interaction data relating to overlapped genes, and the PPI network were acquired using the STRING database. A network was constructed to represent shared genes, consisting of 74 nodes and 130 edges with a degree of 0.8. The network underwent analysis using Cytoscape software, extracting a key network with the highest score from the STRING network (Figure 6A). The CytoHabba plugin within the Cytoscape software was utilized to identify hub genes. Note that the plugin employs various topological techniques, and in the current study, the degree approach was utilized to identify hub genes. The method used is measured precisely in its identification of crucial proteins, as it relies on the highest degree of connections within the network and subsequently assigns them the highest score (Figure 6B). The key genes identified as significant through analysis in the Cytoscape software were insulin receptor substrate 1 (IRS1), fibroblast growth factor 2 (FGF2), mitogen-activated protein kinase 6 (MAPK6), MAPK1, MAPK3, MAPK8, MAPK9, polo like kinase 1 (PLK1), protein kinase CAMP-activated catalytic subunit alpha (PRKACA), and protein kinase C alpha (PRKCA).
4. Discussion
The present study involved selecting and examining 2 distinct sets of samples. The initial cohort comprised endometrium samples, while the subsequent cohort consisted of trophoblasts that were extracted from women who had experienced RMs and healthy controls. Upon conducting a comprehensive analysis of the data, a comparative analysis was performed between the 2 distinct sample types, identifying 41 common genes between the endometrium and trophoblast.
The problem of RMs remains a significant reproductive concern worldwide and a prevalent complication associated with pregnancy.
To understand the abnormality in the fertility and reproductive system leading to fetal loss, it is vital to examine the mechanisms and factors implicated in the ovarian follicle production process and the implantation of the fetus in the maternal uterus, as well as the disorders of these pathways. The processes are greatly influenced by genetics. While the diagnosis of RMs may be challenging, it can be beneficial for both the medical practitioner and the patient, as it enhances the chance of a successful subsequent pregnancy. Thus, identifying effective factors in this regard may be highly advantageous.
Upon studying the pathways implicated in the functioning of overlapped genes, a considerable proportion of genes exhibiting alterations in expression are subject to regulation within the cell cycle pathway. Disruptions in this pathway are associated with the activation of cellular proliferation and the inhibition of apoptosis, as previously reported (11). Moreover, it has been revealed that cell cycle regulation plays a crucial role in the differentiation process of uterine stromal cells during implantation. The genes responsible for regulating the cell cycle exhibit dysregulation during the secretory phase of the endometrium. Hence, regulating gene expression relating to this process through hormone intervention, such as progesterone or steroids, has been observed to be effective in enhancing the implantation rate (12). Also, one of the findings elucidated in this investigation relates to the function of apoptosis mediated by the involved genes. Comparative analyses between samples with miscarriages and healthy samples have revealed a significant increase in the level of apoptosis in the former group. Subsequent investigations have suggested that increased apoptosis may be one of the factors contributing to RMs (13). Apoptosis has been verified to exist in trophoblast during the initial stages of pregnancy. The processes of implantation, blastocyst growth, endometrial regeneration, and placental structure reconstruction, among others, are intricately linked to apoptosis (14).
The balance between cellular proliferation in placental villi and endometrium and the level of apoptosis is crucial in pregnancy physiology. However, in pathological conditions such as miscarriage, this process is disrupted and influenced, leading to an imbalance. The present study displays the PI3K-Akt signaling pathway, which regulates cellular growth and apoptosis. Dysregulation of this pathway increases proliferation or apoptosis within the endometrium (15). Typically, the PI3K-Akt signaling pathway is downregulated in initiating embryo acceptance by the endometrium. During the proliferative phase, the PI3K-Akt signaling pathway expression is diminished in women with a thin endometrium (16).
Thus, the aforementioned approach may also prove effective in abortion. An additional pathway identified is the metabolic pathway of ketone bodies. Ketone bodies are a class of organic compounds characterized by a carbonyl group in the middle of the carbon chain. They are synthesized in the hepatic tissue through the process of ketogenesis. Ketone bodies are a significant substitute fuel for peripheral tissues, particularly in the absence of glucose. Various factors, including glucagon trigger the induction of lipolysis and ketogenesis, while insulin suppresses these processes (17). Research has indicated that pregnant women are at a higher risk of experiencing ketosis than others (18). Numerous investigations, comprising animal model experiments, have demonstrated that environments characterized by elevated ketone levels may result in unfavorable outcomes for both the mother and the fetus (19). The study found that beta-hydroxy-butyric acid exposure in mouse embryos resulted in a range of abnormalities, including growth retardation and neural tube defects. The severity of these abnormalities was found to increase with higher exposure doses (20).
The pathway associated with the activation of the complement system was highly significant among the pathways linked to shared genes. The complement system exhibits activity throughout the menstrual cycle, with heightened expression observed during the secretory phase. The molecules of the complement system impact the attachment between the embryo and the endometrium at the location of implantation. The complement protein C1q has a significant function in the implantation process within the endometrium and is also implicated in the pathogenesis of pre-eclampsia, as evidenced by previous research (21). It have demonstrated that the coagulation and complement cascade pathway is significant in the endometrium during pregnancy (22). Apart from preserving the endometrium through the preservation of epithelial integrity, the presence of a functional complement system also raises worries regarding the possibility of the fetus being perceived as a foreign object. Thus, it is crucial to establish balance in this case (23).
Furthermore, hub genes were identified in the pathways discovered during the study. IRS1 and FGF2 were identified as common genes with the highest score. The IRS1 gene encodes for the insulin receptor and initiates the insulin-dependent signaling cascade. Insulin is a crucial metabolic hormone in maintaining energy homeostasis within the human body (24). The insulin-dependent signaling pathway is a crucial factor in the development of the fetus. The insulin hormone, acting as a growth factor, can potentially enhance cell proliferation and inhibit apoptosis via the PI3K-Akt pathways in endometrial cancer (25). The invasion behavior of endometrial cancer is associated with the activation of the insulin receptor IRS1. Over the initial 20 wk of development, there is a steady increase in fetal growth hormone levels, which significantly impacts insulin metabolism (26). Hence, the balance of energy can impact the embryo implantation process and subsequent growth. The current study demonstrates the reduction in the insulin receptor gene IRS1 expression among females who experienced miscarriages. The FGF2 gene is responsible for encoding the fibroblast growth factor- biomolecules that induce cellular growth, proliferation, and homeostatic regulation. These factors are crucial in regulating proliferation, differentiation, angiogenesis, tissue regeneration, and embryo development (27).
The expression of FGF2 is observed to be elevated in the endometrium during the gestational period. The literature indicates that FGF2 has demonstrated the ability to induce trophoblast migration, adhesion, and embryogenesis across multiple species (28). In addition, it has been established that FGF2, in conjunction with vascular endothelial growth factor, plays a crucial role in regulating the angiogenesis process during placental development (29). The observed reduction in FGF2 expression in samples associated with RMs highlights this factor's significance in maintaining fetal viability throughout gestation.
5. Conclusion
The current study aimed to employ bioinformatics methodologies and tools to identify significant genes and their associated pathways in endometrium and trophoblasts. Furthermore, a comparative analysis was conducted to identify the common genes between the 2 groups by examining the genes exhibiting alterations in expression across both tissues. The study's findings indicate that the genes analyzed in both endometrial and trophoblastic samples are significant in women with RMs compared to those with normal pregnancies. Conversely, the co-occurrence of common genes in the aforementioned groups simultaneously underscores the significance of accuracy in both cell types.
Data availability
The data generated during and/or analyses during the current study are available from the corresponding author upon reasonable request.
Author contributions
K. Ahmadi performed most of the bioinformatics data analysis. S. Reiisi coordinated the study, participated in intellectual discussions of the data, wrote and revised the manuscript. Z. Habibi participated in intellectual discussions of the data and helped in reviewing the manuscript. All authors approved the final manuscript and take responsibility for the integrity of the data.
Acknowledgements
We acknowledged all the individuals who helped do this study, particularly the Research Vice-Chancellor for Research and Innovation of Shahrekord University, Shehrekord, Iran. Artificial intelligence was used to improve the language and readability of the paper.
Conflict of Interest
The authors declare that they have no conflict of interest.
Type of Study: Original Article |
Subject:
Reproductive Genetics
References
1. Practice Committee of the American Society for Reproductive Medicine. Definitions of infertility and recurrent pregnancy loss: A committee opinion. Fertil Steril 2020; 113: 533-535. [DOI:10.1016/j.fertnstert.2019.11.025]
2. Garrido-Gimenez C, Alijotas-Reig J. Recurrent miscarriage: Causes, evaluation and management. Postgrad Med J 2015; 91: 151-162. [DOI:10.1136/postgradmedj-2014-132672]
3. Chen X, Guo D-Y, Yin T-L, Yang J. Non-coding RNAs regulate placental trophoblast function and participate in recurrent abortion. Front Pharmacol 2021; 12: 646521. [DOI:10.3389/fphar.2021.646521]
4. Pollheimer J, Vondra S, Baltayeva J, Beristain AG, Knöfler M. Regulation of placental extravillous trophoblasts by the maternal uterine environment. Front Immunol 2018; 9: 2597. [DOI:10.3389/fimmu.2018.02597]
5. Wu L, Cheng B, Liu Q, Jiang P, Yang J. CRY2 suppresses trophoblast migration and invasion in recurrent spontaneous abortion. J Biochem 2020; 167: 79-87. [DOI:10.1093/jb/mvz076]
6. Mayhew TM. A stereological perspective on placental morphology in normal and complicated pregnancies. J Anat 2009; 215: 77-90. [DOI:10.1111/j.1469-7580.2008.00994.x]
7. Renjith K, Roy DRD, Anthony DJA, Varghese ACV, Sreekutty M, Varma PRVR, et al. Genetic aspects of implantation failure. J Adv Zoology 2023; 44: 498-512.
8. Neykova K, Tosto V, Giardina I, Tsibizova V, Vakrilov G. Endometrial receptivity and pregnancy outcome. J Matern-Fetal Neonatal Med 2022; 35: 2591-2605. [DOI:10.1080/14767058.2020.1787977]
9. Walker ER, McGrane M, Aplin JD, Brison DR, Ruane PT. A systematic review of transcriptomic studies of the human endometrium reveals inconsistently reported differentially expressed genes. Reprod Fertil 2023; 4: e220115. [DOI:10.1530/RAF-22-0115]
10. Bastu E, Demiral I, Gunel T, Ulgen E, Gumusoglu E, Hosseini MK, et al. Potential marker pathways in the endometrium that may cause recurrent implantation failure. Reprod Sci 2019; 26: 879-890. [DOI:10.1177/1933719118792104]
11. Deryabin P, Borodkina A. Stromal cell senescence contributes to impaired endometrial decidualization and defective interaction with trophoblast cells. Hum Reprod 2022; 37: 1505-1524. [DOI:10.1093/humrep/deac112]
12. Pathare ADS, Hinduja I. Endometrial expression of cell adhesion genes in recurrent implantation failure patients in ongoing IVF cycle. Reprod Sci 2022; 29: 513-523. [DOI:10.1007/s43032-021-00708-x]
13. Erboga M, Kanter M. Trophoblast cell proliferation and apoptosis in placental development during early gestation period in rats. Anal Quant Cytopathol Histopathol 2015; 37: 286-294.
14. Zhang X, Wei H. Role of decidual natural killer cells in human pregnancy and related pregnancy complications. Front Immunol 2021; 12: 728291. [DOI:10.3389/fimmu.2021.728291]
15. Crosby D, Glover L, Brennan E, Kelly P, Cormican P, Moran B, et al. Dysregulation of the interleukin-17A pathway in endometrial tissue from women with unexplained infertility affects pregnancy outcome following assisted reproductive treatment. Hum Reprod 2020; 35: 1875-1888. [DOI:10.1093/humrep/deaa111]
16. Makker A, Goel MM, Nigam D, Mahdi AA, Das V, Agarwal A, et al. Aberrant Akt activation during implantation window in infertile women with intramural uterine fibroids. Reprod Sci 2018; 25: 1243-1253. [DOI:10.1177/1933719117737844]
17. Bendridi N, Selmi A, Balcerczyk A, Pirola L. Ketone bodies as metabolites and signalling molecules at the crossroad between inflammation and epigenetic control of cardiometabolic disorders. Int J Mol Sci 2022; 23: 14564. [DOI:10.3390/ijms232314564]
18. Huang J, Yeung AM, Bergenstal RM, Castorino K, Cengiz E, Dhatariya K, et al. Update on measuring ketones. J Diabetes Sci Technol 2024; 18: 714-726. [DOI:10.1177/19322968231152236]
19. Qian M, Wu N, Li L, Yu W, Ouyang H, Liu X, et al. Effect of elevated ketone body on maternal and infant outcome of pregnant women with abnormal glucose metabolism during pregnancy. Diabetes Metab Syndr Obes 2020; 13: 4581-4588. [DOI:10.2147/DMSO.S280851]
20. Whatley EG, Truong TT, Harvey AJ, Gardner DK. Acetoacetate and β-hydroxybutyrate reduce mouse embryo viability via differential metabolic and epigenetic mechanisms. Reprod Biomed Online 2023; 46: 20-33. [DOI:10.1016/j.rbmo.2022.09.018]
21. Agostinis Ch, Mangogna A, Balduit A, Kishore U, Bulla R. A non-redundant role of complement protein C1q in normal and adverse pregnancy. Explor Immunol 2022; 2: 622-636. [DOI:10.37349/ei.2022.00072]
22. Girardi G, Lingo JJ, Fleming SD, Regal JF. Essential role of complement in pregnancy: From implantation to parturition and beyond. Front Immunol 2020; 11: 1681. [DOI:10.3389/fimmu.2020.01681]
23. Spazzapan M, Pegoraro S, Agostinis C, Bulla R. The role of complement component C1q in angiogenesis. Explor Immunol 2023; 3: 574-589. [DOI:10.37349/ei.2023.00122]
24. Laskowski D, Sjunnesson Y, Humblot P, Andersson G, Gustafsson H, Båge R. The functional role of insulin in fertility and embryonic development: What can we learn from the bovine model? Theriogenology 2016; 86: 457-464. [DOI:10.1016/j.theriogenology.2016.04.062]
25. Assaf L, Eid AA, Nassif J. Role of AMPK/mTOR, mitochondria, and ROS in the pathogenesis of endometriosis. Life Sci 2022; 306: 120805. [DOI:10.1016/j.lfs.2022.120805]
26. Tian W, Teng F, Gao J, Gao C, Liu G, Zhang Y, et al. Estrogen and insulin synergistically promote endometrial cancer progression via crosstalk between their receptor signaling pathways. Cancer Biol Med 2019; 16: 55-70. [DOI:10.20892/j.issn.2095-3941.2018.0157]
27. Dejani NN, Nicoletti CF, Argentato PP, Pereira LdS, Saraiva AC, deAssis LM, et al. Maternal plasma transforming growth factor-β1 (TGF-β1) and newborn size: The Araraquara cohort study. J Pediatr 2023; 99: 284-288. [DOI:10.1016/j.jped.2022.11.009]
28. de Ruijter-Villani M, van Boxtel PR, Stout TA. Fibroblast growth factor-2 expression in the preimplantation equine conceptus and endometrium of pregnant and cyclic mares. Theriogenology 2013; 80: 979-989. [DOI:10.1016/j.theriogenology.2013.07.024]
29. Pinto-Bravo P, Rebordão MR, Amaral A, Fernandes C, Galvão A, Silva E, et al. Microvascularization and expression of fibroblast growth factor and vascular endothelial growth factor and their receptors in the mare oviduct. Animals 2021; 11: 1099. [DOI:10.3390/ani11041099]
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
