
International Journal of
Reproductive Biomedicine
Fri, Jul 31, 2026
[Archive]
Volume 3, Issue 1 (7-2005)
IJRM 2005, 3(1): 9-13 |
Back to browse issues page

![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Omrani M D, Nordenskhold A. Mutation detection in human estrogen receptor ? gene in infertile male patients by denaturing high-performance liquid chromatography. IJRM 2005; 3 (1) :9-13
URL: http://ijrm.ir/article-1-33-en.html
URL: http://ijrm.ir/article-1-33-en.html


Full-Text [PDF 113 kb]
(801 Downloads)
| Abstract (HTML) (3422 Views)
This technology, which is based on heteroduplex detection, allows for automated identification of single nucleotide polymorphism (SNP) and small deletions or insertions. Heteroduplex profiles are easily distinguished from homoduplex peaks (8, 9) and thereby provide a reliable means for mutation scanning and discovery. DHPLC has been shown to clearly resolve mutations in various genes with detection rates ranging from 92.5% to 100% (10, 11). Furthermore, it is reported in the literature that the sensitivity of detection by DHPLC is higher than for alternative gel-based analysis techniques; that is, mutations not detected by gel-based techniques could be detected by DHPLC (12, 13).
There is no previous report available which addresses the feasibility of mutation or polymorphism detection by DHPLC analysis for estrogen research β gene (ESR2) gene in infertile male patients. We therefore decided to evaluate the sensitivity and specificity of DHPLC for sequence analysis of the ESR2 gene in these patients. The physiological and pathophysiological roles of estrogens in men have gained increasing attention and are now known to have profound effects on not only the female, but also the male reproductive system. In men, estrogens are synthesized from testosterone mainly in testis, through the action of aromatase cytochrome p450 (14). Estrogen signaling in the cell is mediated by estrogen receptors, of which at least two subtype exist, ESR1 and ESR2. Estrogens, as steroid hormones, act through specific nuclear estrogen receptors (ER), which bind in dimeric form to specific sequences of DNA, in the regulatory region of target genes, and recruit coactivators and corepressors, leading to changes in the rate of transcription of estrogen-regulated genes. These coregulators bind mostly to the ligand-binding domain (15-17). These receptors are present in multiple organ systems, and are important in human development. Two isoforms of the human ER, ESR1 and ESR2, occur, each with distinct tissue and cell patterns of expression, modest overall sequence identity, and with different affinities for different response elements and can therefore yield different transcriptional effects at the same site (18-22).
The recently discovered ESR2 is expressed in both Leydig cells and Sertoli cells in the human testis (23), as well as in most germ cells from spermatogonia to elongating or elongated spermatids. The role of classical ESR1 has been thoroughly studied but there are not many reports of ESR2. Recently, several sequence variants of the ESR2 gene have been described (24). In this study we tried to evaluate the DHPLC analysis for detection of new sequence variations and their relation with male infertility.
As normal control, an unselected group of 96 fertile men, without genital abnormalities and at least one healthy child were chosen. Informed consent was obtained from all subjects or their parents, according to protocol of ethical review board of Uremia University.
Primers were chosen so that an average length of fragments was between 250 and 600 bp (table I).
DHPLC analysis was carried out on an automated HPLC device equipped with a DNA separation column (WAVE: Transgenomic, San Jose, CA, USA) according to the manufacturer’s protocol. Chromatography was performed with buffers A, aqueous solution of 0.1M triethylammonium acetate (TEAA), and B, 0.1M TEAA in 25% acetonitrile (ACN). ACN (Transgenomic), TEAA (Transgenomic), and water used for buffer preparation were of HPLC grade. Eluent conditions for analysis were determined using the Wavemaker™ software (Transgenomic). The eluent flow rate for all analyses was 0.9ml/min. Chromatograms were recorded at a wavelength of 260nm. Analyses under nondenaturing conditions were conducted at 50°C. Mutation detection under partially denaturing conditions was performed at the temperature indicated for each chromatogram.
10ml of each PCR product (containing 10–100ng DNA) was denatured at 95°C for 5 min and then gradually reannealed by decreasing the sample temperature from 95 to 65°C over a period of 30 min. Analysis took in general approximately 7–8 min including column regeneration and re-equilibration to starting conditions. The column mobile phase consisted of a mixture of 0.1M triethylamine acetate (pH 7.0) with (buffer B) or without (buffer A) 25% acetonitrile.
Optimal column temperatures were calculated based upon the melting temperature of the PCR product in either the Transgenomic Wavemaker™ software or in the Stanford melt program (http://www.insertion.stanford.edu/melt.html), as a reference.

Fragments showing an abnormal DHPLC pattern were investigated for identification of sequence variants by automated sequence analysis on the ABI Prism 310. This was performed according to the manufacturer’s protocol using a reamplified PCR product of the abnormal fragment (forward and reverse). The sequence variants were classified according to international databases.
PCR amplicons were purified using the QIAquick PCR Purification Kit (Qiagen, Valencia, Ca.). DNA concentration was determined by spectrophotometry. Cycle sequencing extension products were created in a final volume 20 ml reaction using 12.5ml H2O, 0.5ml forward or reverse primer at 10mg/ml, 0.5ml template DNA, 4ml Big Dye Terminator Ready Reaction mix V1.1 (PE-ABI, Foster City, Ca.) and 2ml 5X Big dye buffer. Cycle sequencing conditions consisted of an initial denaturation step at 96°C for 1 min. followed by 25 cycles of 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min. Unincorporated dye and other contaminants were removed with ethanol precipitation procedure. Data was analyzed with the Sequencers software (Genecodes Inc. Ann Arbor, Michigan).
Statistical analysis
The distributions of ER polymorphisms were compared between the patient group and controls using Fisher's exact test. All statistical tests were two sided. p<0.05 was considered statistically significant.
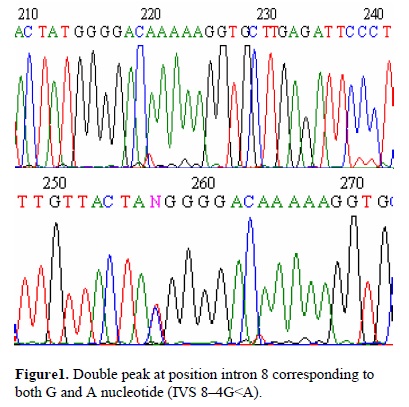
Up to 96 samples can be analyzed in an automated fashion in a single series of runs. About 200 individual samples can be screened for the presence of mutations per day using DHPLC. By applying this method five patients showed heterozygous mutation in intron 8 near intron splicing site (IVS 8–4G<A).
This sequence variation or polymorphism has previously been associated with several conditions, such as bone mineral density in postmenopausal women (25-28), endometrial cancer (29) and systemic blood pressure (30). In addition, estrogens can influence serum androgen levels. This has been recognized in women taking oral contraceptives, or receiving estrogen replacement therapy during menopause, resulting in reduced serum levels of ovarian and adrenal androgens (31-33). Consequently, polymorphisms of the ESR1 and ESR2 were studied in a population-based cohort of 270 women, where long (CA)n polymorphisms have been associated to lower levels of testosterone (15).
Since this variation was not present in the control population with 192 chromosomes in the present study, but it was detected in five of our patients (frequency 5/192=2.6%), therefore it is possible to conclude that there has a statistic significant association between this polymorphism and male infertility in our study (p<0.05).
Yet unable to present any functional explanation for this variation, it is possible that this sequence variation is in linkage disequilibrium with other regulatory sequence variations that may affect gene expression or function, ultimately leading to infertility; or they can cause different structural folds of mRNA and or altered splice, which may possess biological implications, leading to a higher susceptibility to infertility.
Since the polymorphism is located near intron-exon splicing site it could affect the splicing procedure.
Conclusion
In conclusion, the importance of the sequence variation that we’ve found can not yet be understood nor underestimated. Screening the cDNA or, in the case of suspected splice mutations, using accurate models of splice strength, would be needed to predict the consequences of these genetic alterations.
Acknowledgments
We thank the families for their cooperation in the study and also we would like to thank Dr. Ebrahimpour Azar for kindly providing some of the material in this project. This study was funded in part by the Uremia's Medical Science University research deputy.
Full-Text: (483 Views)
Introduction
Techniques for mutation detection in disease-related genes need to be sensitive and specific. Therefore the ideal method to use for mutation analysis particularly if large numbers of DNA fragments need to be analyzed should be sensitive, non-hazardous, relatively inexpensive, and semi- or fully automated to minimize labor and laboratory costs. Denaturing high-performance liquid chromatography (DHPLC) had been shown to meet these criteria for a growing number of applications in disease-related genes, such as CFTR (1), TSC1 (2), hereditary non-poliposis colon cancer (3), mutations in haemochromatosis (4), factor IX and neurofibromatosis type 1 genes (5), BRCA1 and BRCA2 (6, 7).This technology, which is based on heteroduplex detection, allows for automated identification of single nucleotide polymorphism (SNP) and small deletions or insertions. Heteroduplex profiles are easily distinguished from homoduplex peaks (8, 9) and thereby provide a reliable means for mutation scanning and discovery. DHPLC has been shown to clearly resolve mutations in various genes with detection rates ranging from 92.5% to 100% (10, 11). Furthermore, it is reported in the literature that the sensitivity of detection by DHPLC is higher than for alternative gel-based analysis techniques; that is, mutations not detected by gel-based techniques could be detected by DHPLC (12, 13).
There is no previous report available which addresses the feasibility of mutation or polymorphism detection by DHPLC analysis for estrogen research β gene (ESR2) gene in infertile male patients. We therefore decided to evaluate the sensitivity and specificity of DHPLC for sequence analysis of the ESR2 gene in these patients. The physiological and pathophysiological roles of estrogens in men have gained increasing attention and are now known to have profound effects on not only the female, but also the male reproductive system. In men, estrogens are synthesized from testosterone mainly in testis, through the action of aromatase cytochrome p450 (14). Estrogen signaling in the cell is mediated by estrogen receptors, of which at least two subtype exist, ESR1 and ESR2. Estrogens, as steroid hormones, act through specific nuclear estrogen receptors (ER), which bind in dimeric form to specific sequences of DNA, in the regulatory region of target genes, and recruit coactivators and corepressors, leading to changes in the rate of transcription of estrogen-regulated genes. These coregulators bind mostly to the ligand-binding domain (15-17). These receptors are present in multiple organ systems, and are important in human development. Two isoforms of the human ER, ESR1 and ESR2, occur, each with distinct tissue and cell patterns of expression, modest overall sequence identity, and with different affinities for different response elements and can therefore yield different transcriptional effects at the same site (18-22).
The recently discovered ESR2 is expressed in both Leydig cells and Sertoli cells in the human testis (23), as well as in most germ cells from spermatogonia to elongating or elongated spermatids. The role of classical ESR1 has been thoroughly studied but there are not many reports of ESR2. Recently, several sequence variants of the ESR2 gene have been described (24). In this study we tried to evaluate the DHPLC analysis for detection of new sequence variations and their relation with male infertility.
Materials and Methods
Patients
DNA samples were obtained from 96 unrelated infertile men referred to Uremia' University Hospitals presenting with sperm concentrations below 5x106 /ml in at least two ejaculates.As normal control, an unselected group of 96 fertile men, without genital abnormalities and at least one healthy child were chosen. Informed consent was obtained from all subjects or their parents, according to protocol of ethical review board of Uremia University.
PCR amplification
Genomic DNA was extracted from EDTA-preserved blood samples and isolated according to standard procedures (8). Samples used for mutation screening were amplified in 50ml reaction volumes containing approximately 200ng DNA , MgCl2 (25mM) 3ml, dNTP mixture (1.25Mm) 4ml, AmpliTaq GoldTM (5U/ml) 0.5ml, and 50pmol of each primer. In ESR2 exon1, 2 and 3 the amount of MgCl2 (25mM), and dNTP mixture were increased to 4ml and 6ml respectively. Cycle conditions used for amplification were 15 min at 95°C for denaturation, 35 cycles of 30 second at 95°C, 30 second at 56°C and 55 second at 72°C. All amplifications were finished by a prolonged extension step of 4 min at 72°C.Primers were chosen so that an average length of fragments was between 250 and 600 bp (table I).
DHPLC analysis was carried out on an automated HPLC device equipped with a DNA separation column (WAVE: Transgenomic, San Jose, CA, USA) according to the manufacturer’s protocol. Chromatography was performed with buffers A, aqueous solution of 0.1M triethylammonium acetate (TEAA), and B, 0.1M TEAA in 25% acetonitrile (ACN). ACN (Transgenomic), TEAA (Transgenomic), and water used for buffer preparation were of HPLC grade. Eluent conditions for analysis were determined using the Wavemaker™ software (Transgenomic). The eluent flow rate for all analyses was 0.9ml/min. Chromatograms were recorded at a wavelength of 260nm. Analyses under nondenaturing conditions were conducted at 50°C. Mutation detection under partially denaturing conditions was performed at the temperature indicated for each chromatogram.
10ml of each PCR product (containing 10–100ng DNA) was denatured at 95°C for 5 min and then gradually reannealed by decreasing the sample temperature from 95 to 65°C over a period of 30 min. Analysis took in general approximately 7–8 min including column regeneration and re-equilibration to starting conditions. The column mobile phase consisted of a mixture of 0.1M triethylamine acetate (pH 7.0) with (buffer B) or without (buffer A) 25% acetonitrile.
Optimal column temperatures were calculated based upon the melting temperature of the PCR product in either the Transgenomic Wavemaker™ software or in the Stanford melt program (http://www.insertion.stanford.edu/melt.html), as a reference.
Fragments showing an abnormal DHPLC pattern were investigated for identification of sequence variants by automated sequence analysis on the ABI Prism 310. This was performed according to the manufacturer’s protocol using a reamplified PCR product of the abnormal fragment (forward and reverse). The sequence variants were classified according to international databases.
PCR amplicons were purified using the QIAquick PCR Purification Kit (Qiagen, Valencia, Ca.). DNA concentration was determined by spectrophotometry. Cycle sequencing extension products were created in a final volume 20 ml reaction using 12.5ml H2O, 0.5ml forward or reverse primer at 10mg/ml, 0.5ml template DNA, 4ml Big Dye Terminator Ready Reaction mix V1.1 (PE-ABI, Foster City, Ca.) and 2ml 5X Big dye buffer. Cycle sequencing conditions consisted of an initial denaturation step at 96°C for 1 min. followed by 25 cycles of 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min. Unincorporated dye and other contaminants were removed with ethanol precipitation procedure. Data was analyzed with the Sequencers software (Genecodes Inc. Ann Arbor, Michigan).
Statistical analysis
The distributions of ER polymorphisms were compared between the patient group and controls using Fisher's exact test. All statistical tests were two sided. p<0.05 was considered statistically significant.
Results
DHPLC evaluation of 96 infertile patients' (8 with testis degeneration) ESR2 gene, revealed one sequence variations in five patients. They all had double peaks or an atypical fragment configuration indicating heteroduplex formation. For patients with double peaks or even tetra peaks in DHPLC evaluation, sequence variations could be detected. All the five patients showed heterozygous mutation in intron 8 near intron splicing site (IVS 8–4G<A) (Figure1). The matched control population of 96 individuals presented only the normal variant. The difference between the 2 groups was statistical significant (p=0.021).
Discussion
Mutation detection by DHPLC, as it is presented in this context, is a high-throughput, time saving, and economical tool for mutation screening. DHPLC provides information about whether a mutation is present. In order to determine the specific nature of the mutation, however, PCR products in question need to be sequenced.Up to 96 samples can be analyzed in an automated fashion in a single series of runs. About 200 individual samples can be screened for the presence of mutations per day using DHPLC. By applying this method five patients showed heterozygous mutation in intron 8 near intron splicing site (IVS 8–4G<A).
This sequence variation or polymorphism has previously been associated with several conditions, such as bone mineral density in postmenopausal women (25-28), endometrial cancer (29) and systemic blood pressure (30). In addition, estrogens can influence serum androgen levels. This has been recognized in women taking oral contraceptives, or receiving estrogen replacement therapy during menopause, resulting in reduced serum levels of ovarian and adrenal androgens (31-33). Consequently, polymorphisms of the ESR1 and ESR2 were studied in a population-based cohort of 270 women, where long (CA)n polymorphisms have been associated to lower levels of testosterone (15).
Since this variation was not present in the control population with 192 chromosomes in the present study, but it was detected in five of our patients (frequency 5/192=2.6%), therefore it is possible to conclude that there has a statistic significant association between this polymorphism and male infertility in our study (p<0.05).
Yet unable to present any functional explanation for this variation, it is possible that this sequence variation is in linkage disequilibrium with other regulatory sequence variations that may affect gene expression or function, ultimately leading to infertility; or they can cause different structural folds of mRNA and or altered splice, which may possess biological implications, leading to a higher susceptibility to infertility.
Since the polymorphism is located near intron-exon splicing site it could affect the splicing procedure.
Conclusion
In conclusion, the importance of the sequence variation that we’ve found can not yet be understood nor underestimated. Screening the cDNA or, in the case of suspected splice mutations, using accurate models of splice strength, would be needed to predict the consequences of these genetic alterations.
Acknowledgments
We thank the families for their cooperation in the study and also we would like to thank Dr. Ebrahimpour Azar for kindly providing some of the material in this project. This study was funded in part by the Uremia's Medical Science University research deputy.
Type of Study: Original Article |
References
1. Le Marechal C, Audrezet MP, Quere I, Raguenes O, Langonne S, Ferec C. Complete and rapid scanning of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by denaturing high-performance liquid chromatography (DHPLC): major implications for genetic counselling. Hum Genet 2001; 108: 290-298. [DOI:10.1007/s004390100490] [PMID]
2. Roberts PS, Jozwiak S, Kwiatkowski DJ, Dabora SL. Denaturing high-performance liquid chromatography (DHPLC) is a highly sensitive, semi-automated method for identifying mutations in the TSC1 gene. J Biochem Biophys Methods 2001; 47: 33-37. [DOI:10.1016/S0165-022X(00)00149-4]
3. Holinski-Feder E, Muller-Koch Y, Friedl W, Moeslein G, Keller G, Plaschke J, et al. DHPLC mutation analysis of the hereditary nonpolyposis colon cancer (HNPCC) genes hMLH1 and hMSH2. J Biochem Biophys Methods 2001; 47: 21-32. [DOI:10.1016/S0165-022X(00)00148-2]
4. Kaler SG, Devaney JM, Pettit EL, Kirshman R, Marino MA. Novel method for molecular detection of the two common hereditary hemochromatosis mutations. Genet Test 2000; 4: 125-129. [DOI:10.1089/10906570050114821] [PMID]
5. O'Donovan MC, Oefner PJ, Roberts SC, Austin J, Hoogendoorn B, Guy C, et al. Upadhyaya M, Sommer SS, McGuffin P. Blind analysis of denaturing high-performance liquid chromatography as a tool for mutation detection. Genomics 1998; 52: 4-49. [DOI:10.1006/geno.1998.5411] [PMID]
6. Wagner TM, Moslinger RA, Muhr D, Langbauer G, Hirtenlehner K, Concin H, et al. BRCA1-related breast cancer in Austrian breast and ovarian cancer families: specific BRCA1 mutations and pathological characteristics. Int J Cancer 1998; 77: 354-360.
https://doi.org/10.1002/(SICI)1097-0215(19980729)77:3<354::AID-IJC8>3.0.CO;2-N [DOI:10.1002/(SICI)1097-0215(19980729)77:33.0.CO;2-N]
7. Wagner TM, Hirtenlehner K, Shen P, Moeslinger R, Muhr D, Fleischmann E, et al. Lobal sequence diversity of BRCA2: analysis of 71 breast cancer families and 95 control individuals of worldwide populations. Hum Mol Genet 1999; 8: 413-423. [DOI:10.1093/hmg/8.3.413] [PMID]
8. Underhill PA, Jin L, Lin AA . Detection of numerous Y chromosome bialleleic polymorphisms by denaturing high-performance liquid chromatography. Genome Res 1997; 7: 996-1005. [DOI:10.1101/gr.7.10.996] [PMID] [PMCID]
9. Kuklin A, Munson K, Gjerde D, Haefele R, Taylor P. Detection of single-nucleotide polymorphisms with the WAVETM DNA Fragment Analysis System. Genet Test 1997; 1: 201-206. [DOI:10.1089/gte.1997.1.201] [PMID]
10. Liu W, Smith DI, Rechtzigel KJ, Thibodeau SN, James DC. Denaturing high performance liquid chromatography (DHPLC) used in the detection of germline and somatic mutations. Nucleic Acids Res 1998; 26: 1396-1400. [DOI:10.1093/nar/26.6.1396] [PMID] [PMCID]
11. Choy YS, Dabora SL, Hall F, Ramesh V, Niida Y, Franz D, et al. Superiority of denaturing high performance liquid chromatography over single-stranded conformation and conformation-sensitive gel electrophoresis for mutation detection in TSC2. Ann Hum Genet 1999; 63: 383-391. [DOI:10.1046/j.1469-1809.1999.6350383.x] [PMID]
12. Gross E, Arnold N, Goette J, Schwarz-Boeger U, Kiechle M. A comparison of BRCA1 mutation analysis by direct sequencing, SSCP and DHPLC. Hum Gene 1999; 105: 72-78. [DOI:10.1007/s004399900092] [PMID]
13. Wagner T, Stoppa-Lyonnet D, Fleischmann E.Denaturing high-performance liquid chromatography detects reliably BRCA1 and BRCA2 mutations. Genomics 1999; 62: 369-376. [DOI:10.1006/geno.1999.6026] [PMID]
14. Carreau S, Lambard S, Delalande C, Denis-Galeraud I, Bilinska B, Bourguiba S. Aromatase expression and role of estrogens in male gonad. Rev Reprod Bio and Endo 2003; 1:35. [DOI:10.1186/1477-7827-1-35] [PMID] [PMCID]
15. Westberg L, Baghaei F, Rosmond R, Hellstrand M, Landén M, Jansson M, et al. Polymorphisms of the Androgen Receptor Gene and the Estrogen Receptor ß Gene Are Associated with Androgen Levels in Women. J Clin Endocrinol Metab 2001; 86(6): 2562-2568. [DOI:10.1210/jc.86.6.2562]
16. Kong EH, Pike AC, Hubbard RE. Structure and mechanism of the estrogen receptor. Biochem Soc Trans 2003; 31(1): 56-59. [DOI:10.1042/bst0310056] [PMID]
17. Barkhem T, Nilsson S, Gustafsson JA. Molecular mechanisms, physiological consequences and pharmacological implications of estrogen receptor action. Am J Pharmacogenomics 2004; 4(1): 19-28. [DOI:10.2165/00129785-200404010-00003] [PMID]
18. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA 1996; 93: 592-593. [DOI:10.1073/pnas.93.12.5925] [PMID] [PMCID]
19. Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, et al. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab 1997; 82 (12): 4258-4265. [DOI:10.1210/jcem.82.12.4470] [PMID]
20. Gruber CJ, Gruber DM, Gruber IML, Wieser F, Huber JC. Anatomy of the estrogen response element. Trends Endocrinol Metab 2004; 15 (2): 73-78. [DOI:10.1016/j.tem.2004.01.008] [PMID]
21. Warner M, and Gustafsson JA. How to understand estrogen signaling from the phenotypes of ERalpha and ERbeta knockout mice. Ernst Schering Res Found Workshop 2004; 46: 63-77. [DOI:10.1007/978-3-662-05386-7_5] [PMID]
22. Nilsson M, Dahlman-Wright K, Gustafsson JA. Nuclear receptors in disease: the oestrogen receptors. Essays Biochem 2004; 40: 157-167. [DOI:10.1042/bse0400157] [PMID]
23. Chieffi P, Colucci D'Amato L, Guarino F, Salvatore G, Angelini F. 17-estradiol induces spermatogonial proliferation through mitogen-activated protein kinase (extracellular signal-regulated kinase 1/2) activity in the lizard (Podarcis s. sicula), Mol Reprod Dev 2002; 61(2) 218-225. [DOI:10.1002/mrd.1151] [PMID]
24. Elaine CM, Alba MS, Fabiane PT, Francisco JB, Sampaio CFR. Effects of Malnutrition in the Testis. Braz J Urol 2001; 27: 500-506.
25. Scariano JK, Simplicio SG, Montoya GD, Garry PJ, Baumgartner RN. Estrogen receptor beta dinucleotide (CA) repeat polymorphism is significantly associated with bone mineral density in postmenopausal women. Calcif Tissue Int 2004; 74(6): 501-508. [DOI:10.1007/s00223-003-0170-x] [PMID]
26. Shearman AM, Karasik D, Gruenthal KM, Demissie S, Cupples LA, Housman DE, et al. Estrogen receptor beta polymorphisms are associated with bone mass in women and men: the Framingham Study. J Bone Miner Res 2004; 19(5): 773-781. [DOI:10.1359/jbmr.0301258] [PMID]
27. Lau EM, Young RP, Lam V. Estrogen receptor beta gene polymorphisms are associated with higher bone mineral density in premenopausal, but not postmenopausal southern Chinese women. Bone 2002; 31(2): 276-281. [DOI:10.1016/S8756-3282(02)00827-X]
28. Ogawa S, Hosoi T, Shiraki M, Orimo H, Emi M, Muramatsu M, et al. Association of estrogen receptor beta gene polymorphism with bone mineral density. Biochem Biophys Res Commun 2000; 16; 269(2): 537-541. [DOI:10.1006/bbrc.2000.2285] [PMID]
29. Setiawan VW, Hankinson S, Colditz G, Hunter D, Vivo ID. Estrogen receptor beta (ESR2) polymorphisms and endometrial cancer (United States). Cancer Causes Control 2004; 15(6): 627-633. [DOI:10.1023/B:CACO.0000036170.28502.5f] [PMID]
30. Ogawa S, Emi M, Shiraki M, Hosoi T, Ouchi Y, Inoue S. Association of estrogen receptor beta (ESR2) gene polymorphism with blood pressure. J Hum Genet 2000; 45(6):327-330. [DOI:10.1007/s100380070002] [PMID]
31. Tazuke S, Khaw KT, Barrett-Connor E. Exogenous estrogen and endogenous sex hormones. Medicine1992; 71: 44-51. [DOI:10.1097/00005792-199201000-00004] [PMID]
32. Casson PR , Elkind-Hirsch, KE , Buster JE, Hornsby PJ, Carson SA, Snabes MC. Effect of postmenopausal estrogen replacement on circulating androgens. Obstet Gynecol 1997; 90: 995-998. [DOI:10.1016/S0029-7844(97)00538-3]
33. Carr BR, Breslau NA, Givens C, Byrd W, Barnett-Hamm C, Marshburn PB. Oral contraceptive pills, gonadotropin-releasing hormone agonists, or use in combination for treatment of hirsutism: a clinical research center study. J Clin Endocrinol Metab 1995; 80: 1169 -1178. [DOI:10.1210/jcem.80.4.7714086] [PMID]
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
