
International Journal of
Reproductive Biomedicine
Sat, Aug 8, 2026
[Archive]
Volume 5, Issue 4 (7-2007)
IJRM 2007, 5(4): 109-115 |
Back to browse issues page

Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Koruji M, Movahedin M, Mowla S J, Gourabi H. Colony formation ability of frozen thawed spermatogonial stem cell from adult mouse. IJRM 2007; 5 (4) :109-115
URL: http://ijrm.ir/article-1-81-en.html
URL: http://ijrm.ir/article-1-81-en.html


1- Department of Anatomical Sciences, School of Medical Sciences, Tarbiat Modares University, Tehran, Iran
2- Department of Anatomical Sciences, School of Medical Sciences, Tarbiat Modares University, Tehran, Iran ,mansoure@modares.ac.ir
3- Department of Genetics, School of Basic Sciences, Tarbiat Modares University, Tehran, Iran
4- Department of Genetics, Institute of Royan, Tehran, Iran
2- Department of Anatomical Sciences, School of Medical Sciences, Tarbiat Modares University, Tehran, Iran ,
3- Department of Genetics, School of Basic Sciences, Tarbiat Modares University, Tehran, Iran
4- Department of Genetics, Institute of Royan, Tehran, Iran
Full-Text [PDF 215 kb]
(940 Downloads)
| Abstract (HTML) (4116 Views)
Full-Text: (619 Views)
Introduction
Spermatogenesis is a highly organized, complex process that relies on self-replication of undifferentiated SSCs and production of differentiated daughter cells to provide a continual supply of spermatozoa (1). Stem cells are generally defined as colonogenic cells capable of both self- renewal and differentiation in vivo (2, 3). In the adult mouse testis, there are about 35.000 stem cells which is 0.03% of all germ cells (4). However, when spermatogonial cells are cultured in suitable condition, some SSCs survive long-term culture spermatogonial and repopulate recipient testes after transplantation (5).
Antimitotic chemotherapy or radiotherapy can induce failure spermatogenesis. Clinically, spermatogonial stem cell culture and cryopreservation in combination with auto transplantation could serve to restore male fertility after an insult to the testis. An alternative and probably the best method for long-term preservation of SSCs is cryopreservation in combination with culture. Increasing the number of frozen-thawed adult SSCs in vitro is benefit for success in transplantation specially to rescue fertility of cancer patients (6).
To date, various systems have been set up for spermatogonia of various species. Co-culture with Sertoli cell has been demonstrated to support the survival of isolated SSCs from neonate (7, 8) and adult mice testis (5). Izadyar et al (2003) developed a immature bovine spermatogonial-Sertoli cell co-culture system that allowed survival, proliferation and differentiation up to cells showing characteristics of spermatids, during at least 4 months (9). In these cultures, two types of spermatogonial colonies were formed, one consisting of only single stem cells and a mixed type in which, besides stem cells, also pairs and chains of cells were formed. Although these culture systems supported the survival and proliferation of prepubertal type-A spermatogonia greatly in vivo, proliferation was less frequently reported in adult in vitro.
The objective of the current study was proliferation and enhancement of frozen-thawed SSCs numbers during in vitro culture. To accomplish this objective, we developed a Sertoli cells monolayer for co-culture with frozen-thawed adult mice germ cells. We hypothesized that SSC colony formation could be sustained in this system and it could be enhanced by co-culture with Sertoli cells.
Materials and methods
Experimental animals
Male adult mice (age= 6-8 week; n=20) from the National Medical Research Institute (NMRI), initially derived from original stocks obtained from Razi Laboratory (Tehran, Iran), were maintained under standard conditions with free access to food and water at the Animal Facilities of Tarbiat Modares University (Tehran, Iran). The research was conducted in accordance with the National Research Council guidelines.
Spermatogonial cell collection
Bilateral testes were collected from adult 6-8 weeks old NMRI mice for cell suspension. They were placed on ice and transferred to the laboratory within 10 min. After decapsulation, the testes were minced into small pieces and suspended in Dulbecco’s Modified Eagle medium (DMEM; Gibco, Paisley, UK), supplemented with 13.5 gr/L NaHCO3 (Sigma, St Louis,MO, USA), single-strength non-essential amino acids, 100 IUmL−1 penicillin, 100µgmL−1 streptomycin and 40µgmL−1 gentamycin (all from Gibco). The minced pieces of testis were suspended in DMEM, which contained 0.5 mgmL−1 collagenase/dispase, 0.5 mgmL−1 trypsin and 0.08 mgmL−1 DNase, for 30 min (with shaking and a little pipetting ) at 37°C. All the enzymes were purchased from Sigma. After three washes in DMEM medium and removal of most of the interstitial cells, spermatozoa and some spermatids cells, a second digestion step (45 min at 32°C) was performed in DMEM by adding fresh enzymes to the seminiferous cord fragments. Most of the cell aggregates that remained after this treatment were sheared gently by repeated pipetting with a Pasteur pipette for 5 min. The cells were separated from the remaining tubule fragments by centrifugation at 30g for 2 min at 37°C. After filtration through a 70-µm nylon filter, the collected cells were used for spermatocytes cells isolation. The spermatocytes cells were isolated using a procedure described by van Pelt et al. with some modifications (10). The Sertoli cells were isolated using a procedure described by Scarpino et al. with some modification (11) and overnight differential plating. After the spermatocytes cells and Sertoli cells had been isolated, the spermatogonia that remained in suspension were collected and then cultured or cryopreserved.
Spermatocytes cells isolation by PNA binding
Petri dishes with a diameter of 60 mm were coated with 5 ml, 100µg/ml PNA in PBS+ for at least 1 h at 37°C. Then, dishes were washed three times with DMEM containing 0.5% BSA. The dishes were stored with DMEM containing 5 µg/ml DNase for at least 1h at 37°C. Cells were incubated in these dishes for 1.5 h at 32°C in an atmosphere of 5% CO2 in air. After the binding to PNA, nonbinding cells were collected by repeated washing of the dishes with a pipette.
Sertoli cells isolation by DSA-lectin binding
Sertoli cells were extracted from adult mouse testis following the second enzymatic digestion. Briefly, the Petri dishes with a diameter of 60 mm or flask were coated with a solution of 5µgmL−1 of Datura stramonium agglutinin (DSA; Sigma) in phosphate-buffered saline (PBS) at 37°C for 1h. Then, coated plastic dishes were washed three times with DMEM containing 0.5% BSA (BSA; Sigma). The mixed population of the cells obtained by enzymatic digestion was placed on lectin-coated dishes and incubated for 1 h at 32°C in a humidified atmosphere of 5% CO2 in air. After the incubation, the non-adhering cells were collected by being washed twice with medium. Alternatively, 4 days after the Sertoli cells formed a confluent layer, the Sertoli cells were detached by ethylenediamine tetra acetic acid (EDTA) – trypsin treatment (0.02% EDTA–0.1% trypsin in Ca2+- and Mg-free PBS) for 5 min at 37°C, counted and adjusted to desired densities into petri dish for secondary culture in DMEM at 32°C in the presence of 10% fetal bovine serum (FBS; Gibco). This method helped preparation of the Sertoli cells with more than 95% purity.
Cryopreservation and thawing procedure
The isolated cells were cryopreserved using a procedure described by Izadyar et al. with some modification (12). Immediately after cell isolation, cell viability was assessed. Cell suspensions in 0.5-ml aliquots (6 ×106 cells per mL) were prepared. Then, an equal volume of 2× concentrated freezing medium was added dropwise to the Eppendorf vial containing the cell suspension during a period of 10–15 minutes, and after gently mixing by inverting the vial, a sample was taken for viability assessment. The freezing media were based on DMEM supplemented with 10% (v/v) FCS, 1.4M DMSO and 0.07 M sucrose; For noncontrolled-rate freezing, 1.8-mL cryovials vials (Nunc, Denmark) containing 1.0 mL of cell suspension in freezing medium were placed in an insulated (polystyrene) container at -80°C for at least 1 day and then plunged into liquid nitrogen. The cells were thawed by swirling in 38°C water bath for 2 minutes. The contents of the vial was transferred to a tube and diluted slowly by adding two volumes, dropwise, of DMEM supplemented with 10% FCS. Then, the cells were pooled and centrifuged at 2000 × g for 5 minutes, the supernatant was removed, and the pellet was resuspended in DMEM/FCS. A sample was taken for viability assessment, and the remainders of the cells were used for culture experiments.
Fresh and cryopreserved spermatogonial cells co-culture with sertoli cells
A monolayer of Sertoli cells were used to provide an environment that resembles that in vivo as closely as possible. Adult fresh Sertoli cells were used in a co culture system with cryopreserved or fresh adult mouse germ cells.
After formation a Sertoli cells confluent layer, frozen-thawed spermatogonial cells were cultured in two groups: simple culture (Experimental 1) and co culture with Sertoli cells (Experimental 2). In addition, Fresh cells were considered as control groups: simple culture (control1) and co culture with Sertoli cells (control 2). Cells were grown for 3 weeks. At the end of the first week, the cells were passaged and cultured for two weeks. The diameter and the number of colonies were determined every 7th days during the culture for 3 weeks.
Colony assay
Assay of the spermatogonial-cell-derived colonies was carried out in the 7th days. At the end of the first week, the cells were passaged and colony assay was carried out at 14 and 21 days during culture. An inverted microscope (Zeiss, Germany) was used to determine the number of the colonies and their diameters were being measured using ocular grid which was occupied on the inverted microscope.
Immunohistochemistry for confirmation of sertoli and spermatogonia cells
Vimentin was detected by the procedure which was described by Anway et al. in Sertoli cells (13). For an oct-4 immunohistochemistry of the obtained colonies, the colonies grown on the glass slides were stained. Oct-4 immunohistochemistry has been described as a marker for undifferentiated cells (14). Alkaline phosphatase activity was detected by the procedure of Palombi et al (15).
Statistical analysis
Results are expressed in mean±S.D. The statistical significance between mean values was determined by two and one way analysis variance (Tuky-test) in fresh and cryopreserved spermatogonial cells co-culture with Sertoli cells. p≤0.05 was considered significant.
Results
Isolation and characterization of spermatogonial and Sertoli cells
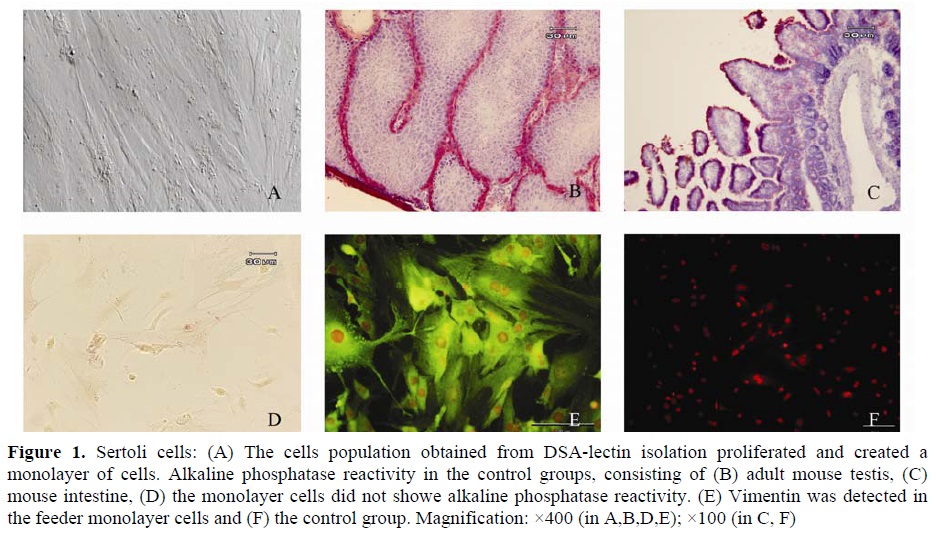
The cells population obtained from DSA-lectin isolation proliferated and created a monolayer of cells (Figure 1.A). They had an irregular outline with a granular appearance. The monolayer cells showed no alkaline phosphatase reactivity (Figure
Effect of cryopreservation on viability rate after thawing
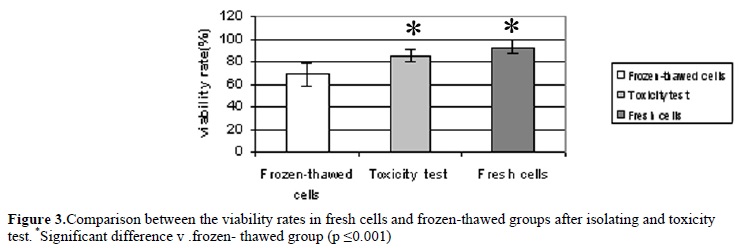
Viability rate of cells after isolation process and after mixing by freezing media were 92.8±6.03% and 85.5±5.7% respectively. These demonstrate that freezing media does not have a significant effect on viability rate. Only 68.4±10.2% of the frozen cells survived after cryopreservation. The viability rate was influenced by the freezing and thawing procedure significantly (p ≤0.001).
Colony assay in fresh and cryopreserved spermatogonial cells co-culture with Sertoli cells
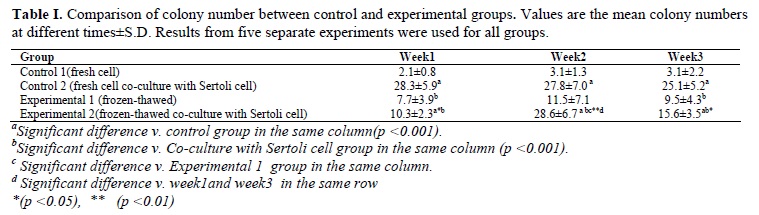
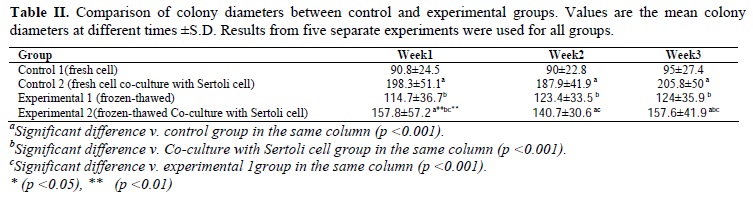
Taken together, as demonstrated in Tables I and II, our results indicated that number of the colonies and their diameters in the co-culture with fresh cells (25.1±5.2 and 205.8±50 µm, respectively) were more than other groups and the differences were significant (p<0.001). Also, number of the colonies and their diameters in experimental 1 (9.5±4.3 and 124±35.9 µm, respectively) and experimental 2 (15.6±3.5 and 157.6±41.9µm, respectively) groups were better than control 1 group (3.1±2.2 and 87.5±30.6µm, respectively) and the differences were significant (p<0.001).
The colonies appeared significantly earlier (on day 4) in co-culture groups compared to colony formation time in control 1 group (on day 7 or after passage).
Discussion
In this study, we demonstrated that co-culture system with Sertoli cells can increase in vitro colony formation of adult fresh and frozen-thawed spermatogonial cells.
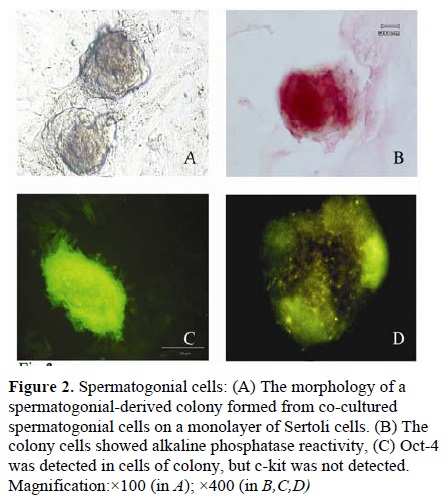
For confirmation of the presence of spermatogonial cells in resulted colonies in addition to alkaline phosphatase activity assessment, c-kit and Oct-4 were traced in the colony cells as well.
Spermatogonial-derived colonies showed alkaline phosphatase activity and Oct-4 expression, but they didn’t showed c-kit expression. This finding is in agreement with that reported by the previous investigators who demonstrated alkaline phosphatase activity and Oct-4 expression in the colony cells (14, 16, 17). But it is in disagreement with Richards et al (1999) who demonstrated primordial germ cells are alkaline phosphatase positive , but from the gonocyte stage onwards, the germ cells are negatively stained (18). In the mouse testis, As and Apr spermatogonia are c-kit-negative, whereas late Aal to B spermatogonia are c-kit positive. Our result in c-kit expression is similar with Schrans-Stassen et al (1999) and Izadyar et al (2002) (19,20). For confirmation of the presence and purification of Sertoli cells as feeder cells, in addition to alkaline phosphatase activity assessment, specific marker detection was carried out using immunocytochemistry anti-Vimentin antibody. Feeder-monolayer did not show alkaline phosphatase activity. Vimentin is a cytoskeletal protein usually found in the epithelial cells; this protein is a marker for Day 14 postnatal Sertoli cells (11) and is strictly localized at the perinuclear region of the cells (21, 22). This finding is in agreement with the reports by Scarpino et al (1998) as well as Anway et al (2003) (11,13).
We demonstrated that frozen/thawed adult spermatogonial stem cells survive from freezing procedure and formed colony in culture. This finding is in agreement with that reported by Izadyar et al (2002) in perpubertal bovine(12). The viability rate of freshly isolated adult spermatogonial cells was higher than that of frozen/thawed cells. Also, the colony formation efficiency of freshly spermatogonial cells co culture with Sertoli cells was higher than that of frozen/thawed cells. This was mainly due to the diminished cell recovery following the freeze/thaw procedure. Reduced cell recovery following the freeze/thaw procedure was also reported by other investigators studying cryopreservation of non pure spermatogonia from other species, including rodents (23, 24) and domestic animals (20,25).
Number of the colonies and their diameters in cryopreserved groups were better than fresh group. Maybe it is due to the damages and shocks resulted from cryopreservation procedure in differentiated germ cells. Cryopreservation probably can causes purification and increases efficiency of colony formation in isolated spermatogonial stem cells.
Number of the colonies in all groups declined at the three weeks. Colonies probably start to develop when spermatogonia and Sertoli cells make contact, apparently creating a microenvironment that favors their development. SSC might need specific micro contacts with niche-offering Sertoli cells during culture (26). Physical contact and secreted growth factors and cytokines affect the survival of spermatogonial stem cells and provide a suitable microenvironment for proliferation and colonies formation. Previous studies have shown that Sertoli cells monolayer support germ cells proliferation and differentiation (27). Better results have been obtained with co-cultures of Sertoli cells with mouse and bovine spermatogonia (9, 26). Other recent results indicate that the presence of Sertoli cells is deleterious to stem cells probably through the induction of differentiation (28). Very likely, the decline may be the result of apoptosis or detachment of differentiating germ cells, as reported previously (26). However, the remaining survived colonies were increased in size. It seems that two week is suitable for duration culture.
Conclusuion
In summary, these findings demonstrate that spermatogonial stem cells from adult mouse testis can be successfully cryopreserved and are able to form colony after freezing and thawing procedures. Also, co-culture system with Sertoli cells can increase in vitro colony formation of adult fresh and frozen-thawed spermatogonial cells.
Acknowledgment
We highly appreciate the contribution of Dr. Ans van Pelt and Prof. Dr. Dirk De Rooij (Fertility Laboratory, AMC, and Amsterdam) for their consults as well as the Shahram Pour biranvand (Department of Anatomical Sciences, School of Medical Sciences, Tarbiat Modares University) and Royan Institute (Tehran, Iran) for its technical assistances. This work was supported by a research grant from Tarbiat Modares University and Iranian Stem cell Network.
Spermatogenesis is a highly organized, complex process that relies on self-replication of undifferentiated SSCs and production of differentiated daughter cells to provide a continual supply of spermatozoa (1). Stem cells are generally defined as colonogenic cells capable of both self- renewal and differentiation in vivo (2, 3). In the adult mouse testis, there are about 35.000 stem cells which is 0.03% of all germ cells (4). However, when spermatogonial cells are cultured in suitable condition, some SSCs survive long-term culture spermatogonial and repopulate recipient testes after transplantation (5).
Antimitotic chemotherapy or radiotherapy can induce failure spermatogenesis. Clinically, spermatogonial stem cell culture and cryopreservation in combination with auto transplantation could serve to restore male fertility after an insult to the testis. An alternative and probably the best method for long-term preservation of SSCs is cryopreservation in combination with culture. Increasing the number of frozen-thawed adult SSCs in vitro is benefit for success in transplantation specially to rescue fertility of cancer patients (6).
To date, various systems have been set up for spermatogonia of various species. Co-culture with Sertoli cell has been demonstrated to support the survival of isolated SSCs from neonate (7, 8) and adult mice testis (5). Izadyar et al (2003) developed a immature bovine spermatogonial-Sertoli cell co-culture system that allowed survival, proliferation and differentiation up to cells showing characteristics of spermatids, during at least 4 months (9). In these cultures, two types of spermatogonial colonies were formed, one consisting of only single stem cells and a mixed type in which, besides stem cells, also pairs and chains of cells were formed. Although these culture systems supported the survival and proliferation of prepubertal type-A spermatogonia greatly in vivo, proliferation was less frequently reported in adult in vitro.
The objective of the current study was proliferation and enhancement of frozen-thawed SSCs numbers during in vitro culture. To accomplish this objective, we developed a Sertoli cells monolayer for co-culture with frozen-thawed adult mice germ cells. We hypothesized that SSC colony formation could be sustained in this system and it could be enhanced by co-culture with Sertoli cells.
Materials and methods
Experimental animals
Male adult mice (age= 6-8 week; n=20) from the National Medical Research Institute (NMRI), initially derived from original stocks obtained from Razi Laboratory (Tehran, Iran), were maintained under standard conditions with free access to food and water at the Animal Facilities of Tarbiat Modares University (Tehran, Iran). The research was conducted in accordance with the National Research Council guidelines.
Spermatogonial cell collection
Bilateral testes were collected from adult 6-8 weeks old NMRI mice for cell suspension. They were placed on ice and transferred to the laboratory within 10 min. After decapsulation, the testes were minced into small pieces and suspended in Dulbecco’s Modified Eagle medium (DMEM; Gibco, Paisley, UK), supplemented with 13.5 gr/L NaHCO3 (Sigma, St Louis,MO, USA), single-strength non-essential amino acids, 100 IUmL−1 penicillin, 100µgmL−1 streptomycin and 40µgmL−1 gentamycin (all from Gibco). The minced pieces of testis were suspended in DMEM, which contained 0.5 mgmL−1 collagenase/dispase, 0.5 mgmL−1 trypsin and 0.08 mgmL−1 DNase, for 30 min (with shaking and a little pipetting ) at 37°C. All the enzymes were purchased from Sigma. After three washes in DMEM medium and removal of most of the interstitial cells, spermatozoa and some spermatids cells, a second digestion step (45 min at 32°C) was performed in DMEM by adding fresh enzymes to the seminiferous cord fragments. Most of the cell aggregates that remained after this treatment were sheared gently by repeated pipetting with a Pasteur pipette for 5 min. The cells were separated from the remaining tubule fragments by centrifugation at 30g for 2 min at 37°C. After filtration through a 70-µm nylon filter, the collected cells were used for spermatocytes cells isolation. The spermatocytes cells were isolated using a procedure described by van Pelt et al. with some modifications (10). The Sertoli cells were isolated using a procedure described by Scarpino et al. with some modification (11) and overnight differential plating. After the spermatocytes cells and Sertoli cells had been isolated, the spermatogonia that remained in suspension were collected and then cultured or cryopreserved.
Spermatocytes cells isolation by PNA binding
Petri dishes with a diameter of 60 mm were coated with 5 ml, 100µg/ml PNA in PBS+ for at least 1 h at 37°C. Then, dishes were washed three times with DMEM containing 0.5% BSA. The dishes were stored with DMEM containing 5 µg/ml DNase for at least 1h at 37°C. Cells were incubated in these dishes for 1.5 h at 32°C in an atmosphere of 5% CO2 in air. After the binding to PNA, nonbinding cells were collected by repeated washing of the dishes with a pipette.
Sertoli cells isolation by DSA-lectin binding
Sertoli cells were extracted from adult mouse testis following the second enzymatic digestion. Briefly, the Petri dishes with a diameter of 60 mm or flask were coated with a solution of 5µgmL−1 of Datura stramonium agglutinin (DSA; Sigma) in phosphate-buffered saline (PBS) at 37°C for 1h. Then, coated plastic dishes were washed three times with DMEM containing 0.5% BSA (BSA; Sigma). The mixed population of the cells obtained by enzymatic digestion was placed on lectin-coated dishes and incubated for 1 h at 32°C in a humidified atmosphere of 5% CO2 in air. After the incubation, the non-adhering cells were collected by being washed twice with medium. Alternatively, 4 days after the Sertoli cells formed a confluent layer, the Sertoli cells were detached by ethylenediamine tetra acetic acid (EDTA) – trypsin treatment (0.02% EDTA–0.1% trypsin in Ca2+- and Mg-free PBS) for 5 min at 37°C, counted and adjusted to desired densities into petri dish for secondary culture in DMEM at 32°C in the presence of 10% fetal bovine serum (FBS; Gibco). This method helped preparation of the Sertoli cells with more than 95% purity.
Cryopreservation and thawing procedure
The isolated cells were cryopreserved using a procedure described by Izadyar et al. with some modification (12). Immediately after cell isolation, cell viability was assessed. Cell suspensions in 0.5-ml aliquots (6 ×106 cells per mL) were prepared. Then, an equal volume of 2× concentrated freezing medium was added dropwise to the Eppendorf vial containing the cell suspension during a period of 10–15 minutes, and after gently mixing by inverting the vial, a sample was taken for viability assessment. The freezing media were based on DMEM supplemented with 10% (v/v) FCS, 1.4M DMSO and 0.07 M sucrose; For noncontrolled-rate freezing, 1.8-mL cryovials vials (Nunc, Denmark) containing 1.0 mL of cell suspension in freezing medium were placed in an insulated (polystyrene) container at -80°C for at least 1 day and then plunged into liquid nitrogen. The cells were thawed by swirling in 38°C water bath for 2 minutes. The contents of the vial was transferred to a tube and diluted slowly by adding two volumes, dropwise, of DMEM supplemented with 10% FCS. Then, the cells were pooled and centrifuged at 2000 × g for 5 minutes, the supernatant was removed, and the pellet was resuspended in DMEM/FCS. A sample was taken for viability assessment, and the remainders of the cells were used for culture experiments.
Fresh and cryopreserved spermatogonial cells co-culture with sertoli cells
A monolayer of Sertoli cells were used to provide an environment that resembles that in vivo as closely as possible. Adult fresh Sertoli cells were used in a co culture system with cryopreserved or fresh adult mouse germ cells.
After formation a Sertoli cells confluent layer, frozen-thawed spermatogonial cells were cultured in two groups: simple culture (Experimental 1) and co culture with Sertoli cells (Experimental 2). In addition, Fresh cells were considered as control groups: simple culture (control1) and co culture with Sertoli cells (control 2). Cells were grown for 3 weeks. At the end of the first week, the cells were passaged and cultured for two weeks. The diameter and the number of colonies were determined every 7th days during the culture for 3 weeks.
Colony assay
Assay of the spermatogonial-cell-derived colonies was carried out in the 7th days. At the end of the first week, the cells were passaged and colony assay was carried out at 14 and 21 days during culture. An inverted microscope (Zeiss, Germany) was used to determine the number of the colonies and their diameters were being measured using ocular grid which was occupied on the inverted microscope.
Immunohistochemistry for confirmation of sertoli and spermatogonia cells
Vimentin was detected by the procedure which was described by Anway et al. in Sertoli cells (13). For an oct-4 immunohistochemistry of the obtained colonies, the colonies grown on the glass slides were stained. Oct-4 immunohistochemistry has been described as a marker for undifferentiated cells (14). Alkaline phosphatase activity was detected by the procedure of Palombi et al (15).
Statistical analysis
Results are expressed in mean±S.D. The statistical significance between mean values was determined by two and one way analysis variance (Tuky-test) in fresh and cryopreserved spermatogonial cells co-culture with Sertoli cells. p≤0.05 was considered significant.
Results
Isolation and characterization of spermatogonial and Sertoli cells
The cells population obtained from DSA-lectin isolation proliferated and created a monolayer of cells (Figure 1.A). They had an irregular outline with a granular appearance. The monolayer cells showed no alkaline phosphatase reactivity (Figure
1.D). Meanwhile, the control groups, consisting of mouse intestine (brush border of villous), adult mouse testes (the endothelial cells, smooth muscle cells of the blood capillaries in the interstitium and peritubular myoid cells) showed alkaline phosphatase reactivity (Figure 1B, C). Moreover, Vimentin, which is a molecular marker for Sertoli
cells, was detected in the feeder monolayer cells
(Figure 1E, F). The other cells type, with spherical outline and two or three eccentrically placed nucleoli, created a colony after proliferation during first week or immediately after passage (Figure 2.A). The resulted colonies had clearly AP activity. Moreover, Oct-4, which is a molecular marker for SSCs, was detected in the obtained colonies (Figure 2.C, D) but c-kit wasn’t detected in them.
cells, was detected in the feeder monolayer cells
(Figure 1E, F). The other cells type, with spherical outline and two or three eccentrically placed nucleoli, created a colony after proliferation during first week or immediately after passage (Figure 2.A). The resulted colonies had clearly AP activity. Moreover, Oct-4, which is a molecular marker for SSCs, was detected in the obtained colonies (Figure 2.C, D) but c-kit wasn’t detected in them.
Effect of cryopreservation on viability rate after thawing
Viability rate of cells after isolation process and after mixing by freezing media were 92.8±6.03% and 85.5±5.7% respectively. These demonstrate that freezing media does not have a significant effect on viability rate. Only 68.4±10.2% of the frozen cells survived after cryopreservation. The viability rate was influenced by the freezing and thawing procedure significantly (p ≤0.001).
Colony assay in fresh and cryopreserved spermatogonial cells co-culture with Sertoli cells
Taken together, as demonstrated in Tables I and II, our results indicated that number of the colonies and their diameters in the co-culture with fresh cells (25.1±5.2 and 205.8±50 µm, respectively) were more than other groups and the differences were significant (p<0.001). Also, number of the colonies and their diameters in experimental 1 (9.5±4.3 and 124±35.9 µm, respectively) and experimental 2 (15.6±3.5 and 157.6±41.9µm, respectively) groups were better than control 1 group (3.1±2.2 and 87.5±30.6µm, respectively) and the differences were significant (p<0.001).
The colonies appeared significantly earlier (on day 4) in co-culture groups compared to colony formation time in control 1 group (on day 7 or after passage).
Discussion
In this study, we demonstrated that co-culture system with Sertoli cells can increase in vitro colony formation of adult fresh and frozen-thawed spermatogonial cells.
For confirmation of the presence of spermatogonial cells in resulted colonies in addition to alkaline phosphatase activity assessment, c-kit and Oct-4 were traced in the colony cells as well.
Spermatogonial-derived colonies showed alkaline phosphatase activity and Oct-4 expression, but they didn’t showed c-kit expression. This finding is in agreement with that reported by the previous investigators who demonstrated alkaline phosphatase activity and Oct-4 expression in the colony cells (14, 16, 17). But it is in disagreement with Richards et al (1999) who demonstrated primordial germ cells are alkaline phosphatase positive , but from the gonocyte stage onwards, the germ cells are negatively stained (18). In the mouse testis, As and Apr spermatogonia are c-kit-negative, whereas late Aal to B spermatogonia are c-kit positive. Our result in c-kit expression is similar with Schrans-Stassen et al (1999) and Izadyar et al (2002) (19,20). For confirmation of the presence and purification of Sertoli cells as feeder cells, in addition to alkaline phosphatase activity assessment, specific marker detection was carried out using immunocytochemistry anti-Vimentin antibody. Feeder-monolayer did not show alkaline phosphatase activity. Vimentin is a cytoskeletal protein usually found in the epithelial cells; this protein is a marker for Day 14 postnatal Sertoli cells (11) and is strictly localized at the perinuclear region of the cells (21, 22). This finding is in agreement with the reports by Scarpino et al (1998) as well as Anway et al (2003) (11,13).
We demonstrated that frozen/thawed adult spermatogonial stem cells survive from freezing procedure and formed colony in culture. This finding is in agreement with that reported by Izadyar et al (2002) in perpubertal bovine(12). The viability rate of freshly isolated adult spermatogonial cells was higher than that of frozen/thawed cells. Also, the colony formation efficiency of freshly spermatogonial cells co culture with Sertoli cells was higher than that of frozen/thawed cells. This was mainly due to the diminished cell recovery following the freeze/thaw procedure. Reduced cell recovery following the freeze/thaw procedure was also reported by other investigators studying cryopreservation of non pure spermatogonia from other species, including rodents (23, 24) and domestic animals (20,25).
Number of the colonies and their diameters in cryopreserved groups were better than fresh group. Maybe it is due to the damages and shocks resulted from cryopreservation procedure in differentiated germ cells. Cryopreservation probably can causes purification and increases efficiency of colony formation in isolated spermatogonial stem cells.
Number of the colonies in all groups declined at the three weeks. Colonies probably start to develop when spermatogonia and Sertoli cells make contact, apparently creating a microenvironment that favors their development. SSC might need specific micro contacts with niche-offering Sertoli cells during culture (26). Physical contact and secreted growth factors and cytokines affect the survival of spermatogonial stem cells and provide a suitable microenvironment for proliferation and colonies formation. Previous studies have shown that Sertoli cells monolayer support germ cells proliferation and differentiation (27). Better results have been obtained with co-cultures of Sertoli cells with mouse and bovine spermatogonia (9, 26). Other recent results indicate that the presence of Sertoli cells is deleterious to stem cells probably through the induction of differentiation (28). Very likely, the decline may be the result of apoptosis or detachment of differentiating germ cells, as reported previously (26). However, the remaining survived colonies were increased in size. It seems that two week is suitable for duration culture.
Conclusuion
In summary, these findings demonstrate that spermatogonial stem cells from adult mouse testis can be successfully cryopreserved and are able to form colony after freezing and thawing procedures. Also, co-culture system with Sertoli cells can increase in vitro colony formation of adult fresh and frozen-thawed spermatogonial cells.
Acknowledgment
We highly appreciate the contribution of Dr. Ans van Pelt and Prof. Dr. Dirk De Rooij (Fertility Laboratory, AMC, and Amsterdam) for their consults as well as the Shahram Pour biranvand (Department of Anatomical Sciences, School of Medical Sciences, Tarbiat Modares University) and Royan Institute (Tehran, Iran) for its technical assistances. This work was supported by a research grant from Tarbiat Modares University and Iranian Stem cell Network.
Type of Study: Original Article |
References
1. Oatley JM, de Avila DM, Reeves JJ, McLean DJ. Testis tissue explant culture supports survival and proliferation of bovine spermatogonial stem cells. Biol Reprod 2004; 70: 625-631. [DOI:10.1095/biolreprod.103.022483]
2. Fuchs E, Segre JA. Stem cells: a new lease on life. Cell 2000; 100:143- 155. [DOI:10.1016/S0092-8674(00)81691-8]
3. Weissman IL. Translating stem and progenitor cell biology to the clinic: barriers and opportunities. Science 2000; 287:1442-1446. [DOI:10.1126/science.287.5457.1442]
4. Tegelenbosch RAJ, de Rooij DGA quantitative study of spermatogonial multiplication and stem cell renewal in the C3H: 101 F1 hybrid mouse. Mutat Res 1993; 290, 193-200. [DOI:10.1016/0027-5107(93)90159-D]
5. Nagano M, Brinster R.L. Spermatogonial transplantation and reconstitution of donor cell spermatogenesis in recipient mice, APMIS 1998; 106:47-55. [DOI:10.1111/j.1699-0463.1998.tb01318.x]
6. Izadyar F, Creemers LB, van Dissel-Emiliani FMF, de Rooij DG. Spermatogonial stem cell transplantation. Molecular and Cellular Endocrinology 2000; 169:21-25. [DOI:10.1016/S0303-7207(00)00346-4]
7. Maekawa M, Nishimune Y. In-vitro proliferation of germ cells and supporting cells in t he neonatal mouse testis. Cell Tissue Res 1991; 265: 551-554. [DOI:10.1007/BF00340879]
8. Anjamrooz SH, Movahedin M, Tiraihi T, Mowla SJ. In vitro effects of epidermal growth factor, follicle stimulating hormone and testosterone on mouse spermatogonial cell colony formation.Reproduction, Fertility and Development 2006;18, 709-720. [DOI:10.1071/RD05126]
9. Izadyar F, den Ouden K, Creemers LB, Posthuma G, Parvinen M, de Rooij DG. Proliferation and differentiation of bovine type A spermatogonia during long-term culture. Biol Reprod 2003; 68:272-281. [DOI:10.1095/biolreprod.102.004986]
10. van Pelt AMM, Morena AR, van Dissel-Emiliani FM, Boitani C, Gaemers IC, de Rooij DG, et al. Isolation of the synchronized A spermatogonia from adult vitamin A-deficient rat testes. Biol Reprod 1996; 55: 439-444. [DOI:10.1095/biolreprod55.2.439]
11. Scarpino S, Morena AR, Petersen C, Froysa B, Soder O, Boitani CA. Rapid method of Sertoli cells isolation by DSA lectin, allowing mitotic analyses. Mol Cell Endocrinol 1998; 146: 121-127. [DOI:10.1016/S0303-7207(98)00190-7]
12. Izadyar F, Matthijs-Rijsenbilt JJ, den Ouden K, Creemers LB, Woelders H, de Rooij DG. Development of a cryopreservation protocol for type A spermatogonia. J Androl 2002; 3:537-545.
13. Anway MD, Folmer J, Wright WW, Zirk BR. Isolation of Sertoli cells from Adult Rat Testes: An Approach to Ex Vivo Studies of Sertoli cells Function. Biol Reprod 2003; 68:996-1002 [DOI:10.1095/biolreprod.102.008045]
14. Kubota H, Avarbock MR, Brinster RL. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Prroc Natl Accad Sci USA 2004; 101: 16489-16494. [DOI:10.1073/pnas.0407063101]
15. Palombi F, Dicarlo C. Alkaline Phosphatase is a Marker for Myoid Cells in Cultures of Rat Peritubular and Tubular Tissue. Biol Reprod 1988; 39: 1101-1109. [DOI:10.1095/biolreprod39.5.1101]
16. Jeong D, McLean DJ, Griswold MD. Long-term culture and transplantation of murine testicular germ cells. J Androl 2003; 24: 661-669. [DOI:10.1002/j.1939-4640.2003.tb02724.x]
17. Shi YQ, Wang QZ, Liao SY, Zhang Y, Liu YX, Han CS. In vitro propagation of spermatogonial stem cells from KM mice. Front Bio Sci 2006; 11, 2614-2622. [DOI:10.2741/1995]
18. Richards AJ, Enders GC, Resnick JL. Differentiation of murine premigratory primordial germ cells in culture. Biol Reprod 1999; 61: 1146-1151. [DOI:10.1095/biolreprod61.4.1146]
19. Schrans-Stassen BHGJ, van de Kant HJG, de Rooij DG, van Pelt AMM. Differential expression of c-kit in mouse undifferentiated and differentiating type A spermatogonia. Endocrinology 1999; 140:5894-5900. [DOI:10.1210/endo.140.12.7172]
20. Izadyar F, Spierenberg GT, Creemers LB, den Ouden K, de Rooij DG. Isolation and purificaton of type A spermatogonia from the bovine testis. Reprod 2002; 124:85-94. [DOI:10.1530/rep.0.1240085]
21. Mori C, Nakamura N, Dix DJ, Fujioka M, Nakagawa S, Shiota K, et al. Morphological analysis of germ cell apoptosis during postnatal testis development in normal and Hsp 70-2 knockout mice. Dev Dyn 1997; 208:125-136.
https://doi.org/10.1002/(SICI)1097-0177(199701)208:1<125::AID-AJA12>3.0.CO;2-5 [DOI:10.1002/(SICI)1097-0177(199701)208:13.0.CO;2-5]
22. Oke BO, Suarez-Quian CA. Localization of secretory, membrane-associated and cytoskeletal proteins in rat testis using an improved immunocytochemical protocol that employs polyester wax. Biol Reprod 1993; 48: 621-631. [DOI:10.1095/biolreprod48.3.621]
23. Avarbock MR, Brinster CJ. Brinster RL. Reconstitution of spermatogenesis from frozen spermatogonial stem cells. Nat Med 1996; 2: 693-696. [DOI:10.1038/nm0696-693]
24. Brinster RL, Nagano M. Spermatogonial stem cell transplantation, cryopreservation and culture. Cell Dev Biol 1998; 9: 401-409. [DOI:10.1006/scdb.1998.0205]
25. Dobrinski I, Avarbock MR, Brinster RL. Germ cell transplantation from large domestic animals into mouse testes. Mol Reprod Dev 2000; 56: 270-279.
https://doi.org/10.1002/1098-2795(200011)57:3<270::AID-MRD9>3.0.CO;2-Z [DOI:10.1002/1098-2795(200011)57:33.0.CO;2-Z]
26. Aponte PM, Soda T, van de Kant HJ, de Rooij DG. Basic features of bovine spermatogonial culture and effects of glial cell line-derived neurotrophic factor. Theriogenology 2006; 65: 1828-47. [DOI:10.1016/j.theriogenology.2005.10.020]
27. Creemers LB, den Ouden K, van Pelt AMM, de Rooij DG. Maintenance of adult mouse type A spermatogonia in vitro: influence of serum and growth factors and comparison with prepubertal spermatogonial cell culture. Reproduction 2002; 124: 791-99. [DOI:10.1530/rep.0.1240791]
28. Nagano M, Ryu BY, Brinster CJ, Avarbock MR, Brinster RL. Maintenance of mouse male germ line stem cells in vitro. Biol Reprod 2003; 68:2207-14. [DOI:10.1095/biolreprod.102.014050]
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
