
International Journal of
Reproductive Biomedicine
Thu, Jul 23, 2026
[Archive]
Volume 19, Issue 5 (May 2021)
IJRM 2021, 19(5): 477-482 |
Back to browse issues page



![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Mohammadi R, Taheri R, Shahriyari F, Feiz F, Mohammadi Z, Shirian S, et al . Prenatal diagnosis of de novo small supernumerary marker chromosome 4q (4q11-q12): A case report. IJRM 2021; 19 (5) :477-482
URL: http://ijrm.ir/article-1-1877-en.html
URL: http://ijrm.ir/article-1-1877-en.html
Reza Mohammadi1 

 , Raheleh Taheri1 , Fatemeh Shahriyari1 , Farnaz Feiz2 , Zahra Mohammadi3 , Sadegh Shirian4 , Reza Rraoofian5 , Abdorrasoul Malekpour5 , Reza Pazhoomand *6
, Raheleh Taheri1 , Fatemeh Shahriyari1 , Farnaz Feiz2 , Zahra Mohammadi3 , Sadegh Shirian4 , Reza Rraoofian5 , Abdorrasoul Malekpour5 , Reza Pazhoomand *6
, Raheleh Taheri1 , Fatemeh Shahriyari1 , Farnaz Feiz2 , Zahra Mohammadi3 , Sadegh Shirian4 , Reza Rraoofian5 , Abdorrasoul Malekpour5 , Reza Pazhoomand *6
1- Genetic Laboratory of Shiraz Fertility Center, Shiraz, Iran.
2- Shiraz University of Medical Sciences, Shiraz, Iran.
3- Pathobiology Laboratory of Ordibehesht Hospital, Shiraz, Iran.
4- Department of Pathology, School of Veterinary Medicine, Shahrekord University, Shahrekord, Iran. Shiraz Molecular Pathology Research Center, Dr Daneshbod Pathol Lab, Shiraz, Iran. Shefa Neurosciences Research Center, Tehran, Iran.
5- Legal Medicine Research Center, Legal Medicine Organization, Tehran, Iran.
6- Genetic Laboratory of Shiraz Fertility Center, Shiraz, Iran. Legal Medicine Research Center, Legal Medicine Organization, Tehran, Iran. ,Pazhoomand@yahoo.com
2- Shiraz University of Medical Sciences, Shiraz, Iran.
3- Pathobiology Laboratory of Ordibehesht Hospital, Shiraz, Iran.
4- Department of Pathology, School of Veterinary Medicine, Shahrekord University, Shahrekord, Iran. Shiraz Molecular Pathology Research Center, Dr Daneshbod Pathol Lab, Shiraz, Iran. Shefa Neurosciences Research Center, Tehran, Iran.
5- Legal Medicine Research Center, Legal Medicine Organization, Tehran, Iran.
6- Genetic Laboratory of Shiraz Fertility Center, Shiraz, Iran. Legal Medicine Research Center, Legal Medicine Organization, Tehran, Iran. ,
Full-Text [PDF 1212 kb]
(1331 Downloads)
| Abstract (HTML) (2806 Views)
Full-Text: (626 Views)
- Introduction
Small supernumerary marker chromosomes (sSMCs) are chromosomal fragments with abnormal structures that may not be detected by banding analysis. sSMCs have a size similar to chromosome 20 or smaller and cannot be detected by routine banding pattern analysis (1, 2). While the frequency of prenatal sSMC with no evident origin has been reported as 0.075% and 0.044% in live births, it is 7 times more (i.e., 0.288%) in mentally retarded cases and 0.125% in subfertile cases. About 23% of the cases encompass inherited sSMC which are commonly paternal (16% vs 7%). Worldwide, there are ~2.7x106 living sSMC carriers; 1.8x106 have a de novo sSMC, while ~70% of them are clinically normal (3-5). There are no definite sSMCs karyotype–phenotype correlations, and the phenotypes may range from normal to having dysmorphic features and/or developmental delay, depending on the involved chromosomal region, tissue distribution of the sSMC, and the level of mosaicism (2). Therefore, there is an urgent need for prenatal genetic diagnosis of new sSMCs to forecast the clinical consequences of sSMC and prevention of possible clinical outcomes (2, 6). In this study, we aim to report a rare case of prenatal diagnosed de novo sSMCs derived from the long arm of chromosome 4 [sSMC (4)] using array comparative genomic hybridization (array CGH) technique for the first time.
- Case Report
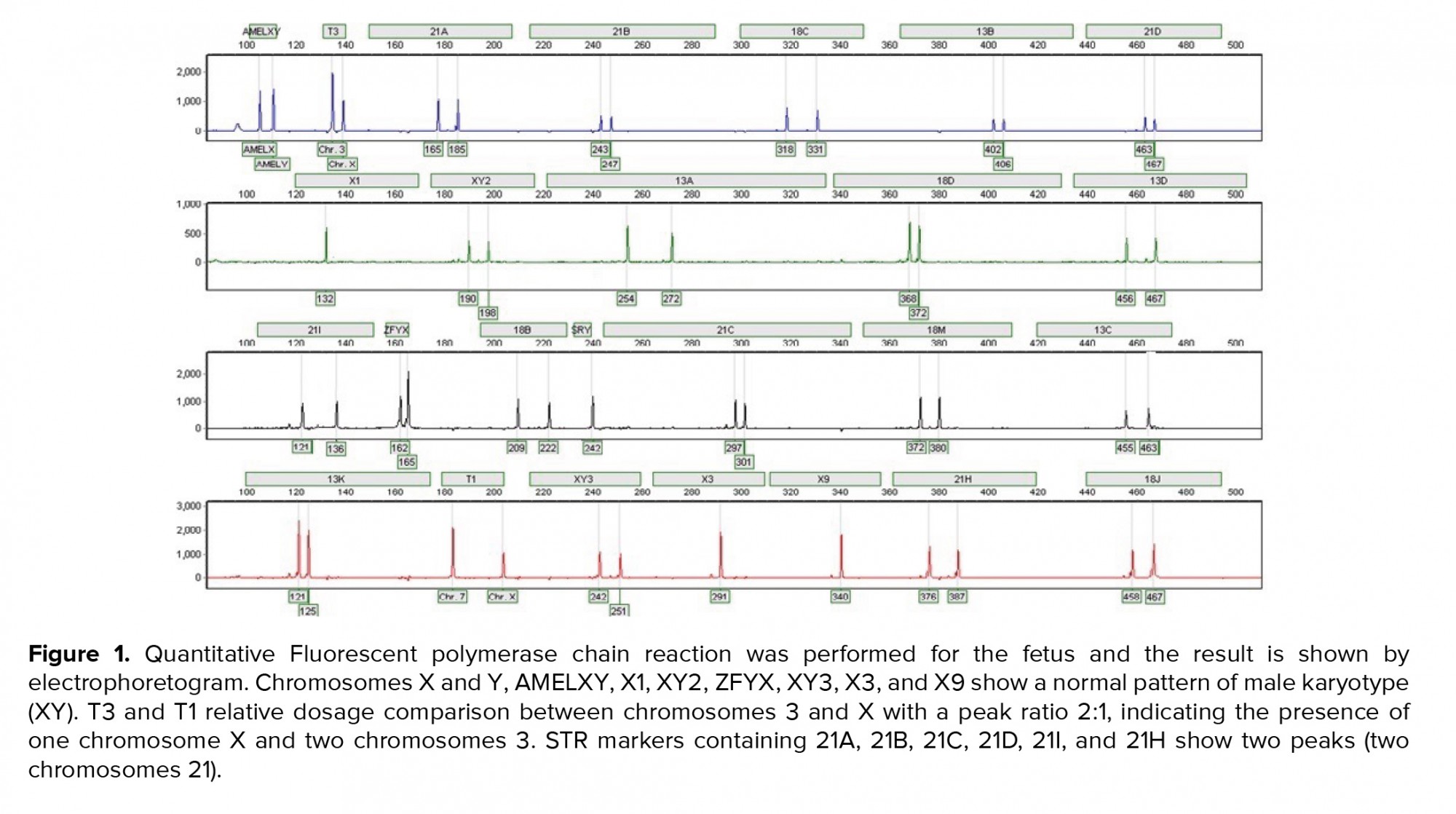
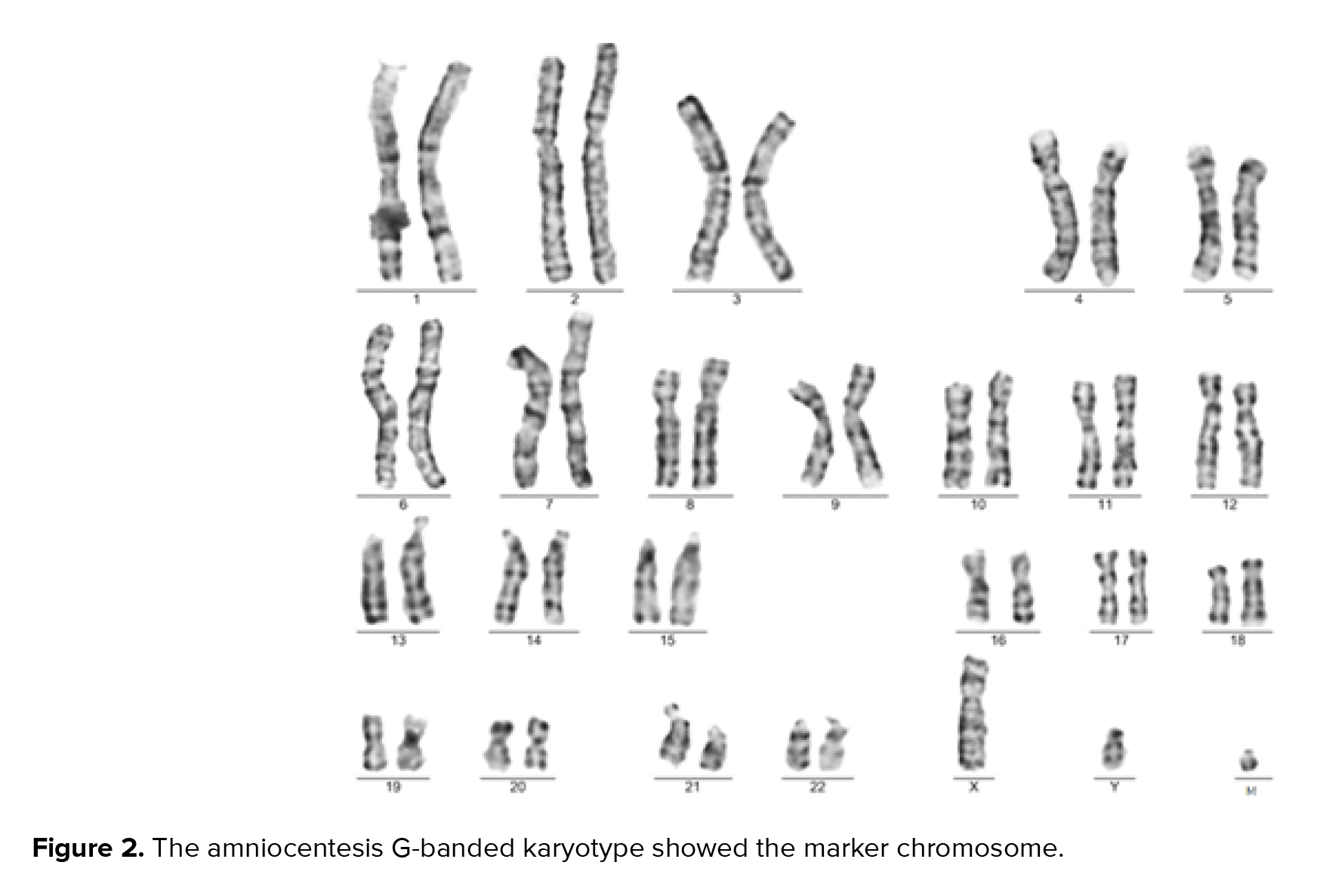
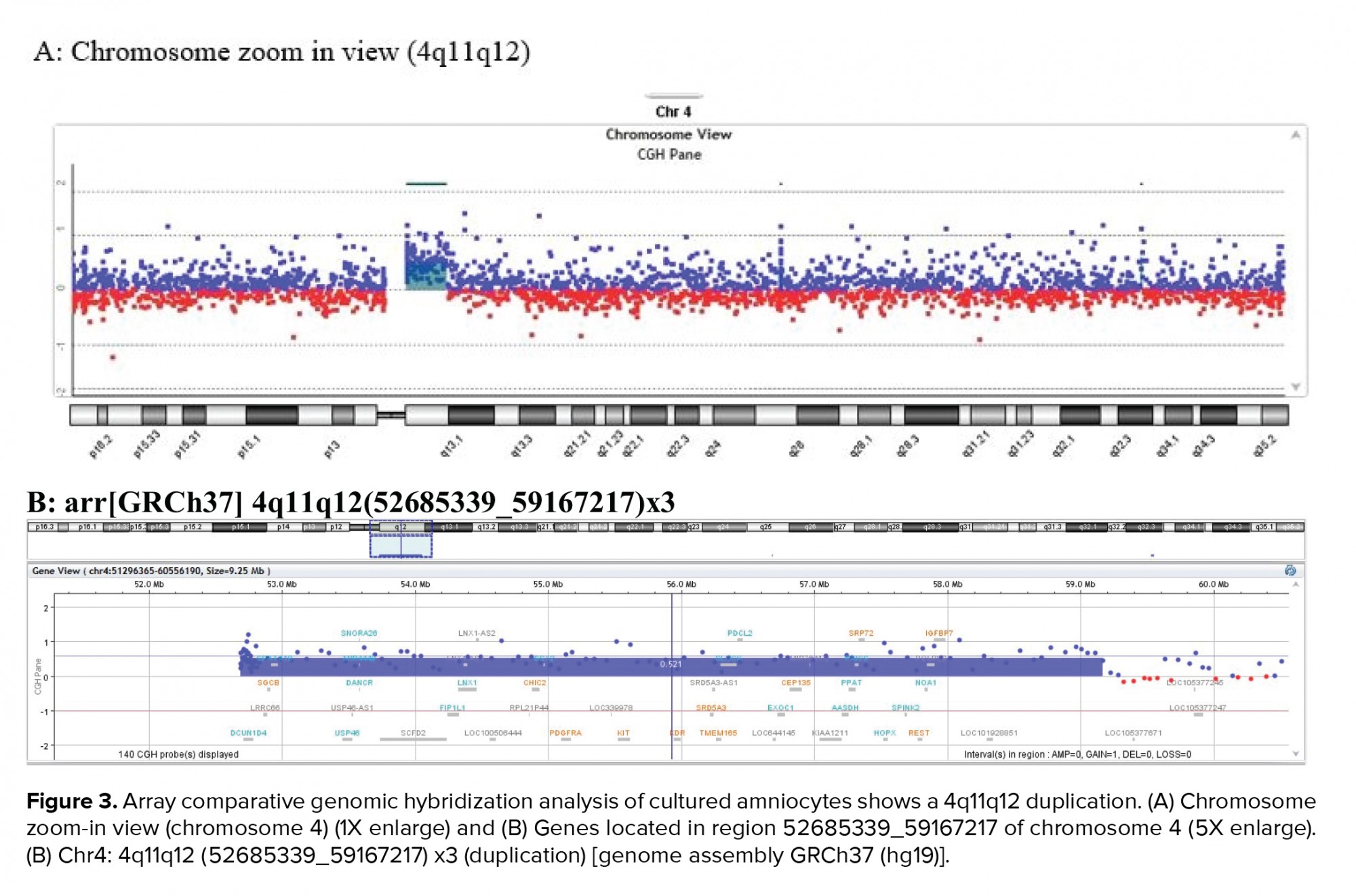
A 36-yr-old surrogate pregnant woman was detected to have a fetus with a high risk of Down’s syndrome (trisomy 21) during the screening test at the 16th wk of gestation. All parameters of the ultrasonography scan at 11 wk and 3 days were normal, the nasal bone was present, and nuchal translucency thickness measured 1.4 mm. The results of biochemical analysis of maternal serum indicated that the risk of Down’s syndrome was higher than that of the screening cut-off (1:30). The parents were phenotypically normal and there was no family history of congenital malformations. The amniocentesis was performed at the 16th wk of the gestation. For the detection of numerical aneuploidy of X, Y, 13, 18, and 21 chromosomes, specific microsatellites were amplified using Quantitative Fluorescent polymerase chain reaction kit (Devyser Compact® v3, Sweden). There was a male fetus, and the electropherogram did not reveal numerical aneuploidy in the mentioned chromosomes (Figure 1). GTG-banding analysis of 100 metaphase spreads showed same-sized sSMC in all primary amniocyte cultures and the fetal karyotype was detected 47,XY,+mar (Figure 2). Karyotype analysis was conducted on the peripheral blood of the biological parents (mother and father were 40 and 45 yr old, respectively) to determine the possible origin of the marker chromosome. There were normal karyotype patterns in all 30 examined cells. Array CGH technique was applied to identify the origin of the sSMC. Whole-genome array CGH was conducted on the DNA extracted from cultured amniocytes using Sure Print G3 ISCA V2 8x60K (Agilent Technologies, Santa Clara, CA, USA). The array comprises of 60,000 spots and identifies 500 established disease regions with the probe spacing median of ≥ 60 Kb. Array CGH analysis on the cultured amniocytes revealed a 6.48-Mb duplication at 4q11q12 (arr [GRCh37] 4q11q12 (52685339_59167217) x3) (Figure 3). The 4q11q12 duplication contains 33 OMIM genes, including 12 disease-causing regions such as SGCB, CHIC2, PDGFRA, KIT, KDR, SRD5A3, TMEM165, CEP135, SRP72, REST, SPINK2, and IGFBP7. Based on the mentioned findings, the parents decided to terminate the pregnancy at 18 wk and 5days of gestation.
2.1. Ethical considerations
The biological parents gave consent for amniocentesis and subsequent analysis and the use of the obtained results for publication.
3. Discussion
The impact of sSMC on prenatal genetic counseling has been a major challenge, is mostly based on theoretical data, and can be improved by the genotype–phenotype correlation studies and molecular cytogenetic analysis in which the chromosomal origins of the sSMC are detected (7). The chromosomal origin of sSMC must be detected to establish a reliable genotype–phenotype correlation (8). In our case, the prenatal molecular cytogenetic assay led to the detection of a de novo sSMCs derived from the proximal region of the long arm of chromosome 4 that resulted in trisomy of 4q11q12. The individuals carrying very small 4q11eq13proximal duplications seem healthy with normal features, however, they may have learning disability and developmental delay (4). However, the duplications of the proximal region of 4q have been contributed to different abnormalities and clinically important features. For example, a 4q12eq13 duplication in a 6-yr-old girl with microcephaly, facial dimorphism clinodactyly of the fifth finger, and psychomotor retardation has been detected (9). A 47,XY,+r [4] (::p10/q12::) karyotype in a 27-yr-old male with facial dimorphism, severe mental retardation, language disability, syndactyly of foot, as well as clinodactyly of the hand has reported (10). A 15-yr-old girl with 4q13.1eq22.2 duplication, who had minor physical anomalies and moderate intellectual disabilities has been reported (11). A 2-yr-and-8-month-old boy with duplication of 4q12eq13 who had microcephaly, mild facial dimorphism, and mental retardation has been previously reported (12). Bonnet and co-workers reported a six-yr-old obese girl with a developmental delay who had 82% mosaicism for an sSMC [4] 4q10eq13 in peripheral lymphocytes (13). An eight-yr-old girl with 8.6-Mb duplication of 4q13.1eq13.3 with developmental delay, attention-deficit hyperactivity, and speaking disability has been previously reported (14). A 47, XX, +mar has been detected with 4p11eq12 sSMC in which only long philtrum and hypertelorism were observed at the termination of pregnancy (15). A three-yr-old boy with 4p11eq12-derived sSMC [4] presenting developmental delay, mild motor retardation, and mild hypotonic features has also been shown (7). As mentioned, de novo sSMCs were not indicated in any of aforementioned studies, and such de novo sSMCs may be undetected causing major clinical manifestations. Our study provides useful information for genetic counseling on prenatally detectable sSMC of 4q11q12.
4. Conclusion
It has been concluded that if the marker chromosome is seen in the amniotic fluid sample but does not appear in parents, the CGH array is needed for making the best decision. Such findings help us in concise genetic counseling and guidance of couples making proper decisions about their fetuses.
Acknowledgements
The authors of this study thank the personnel of the genetic laboratory of Shiraz Fertility Center, Shiraz, Iran, for their kind cooperation.
Conflict of Interest
The authors have no conflicts of interest relevant to this article.
2.1. Ethical considerations
The biological parents gave consent for amniocentesis and subsequent analysis and the use of the obtained results for publication.
3. Discussion
The impact of sSMC on prenatal genetic counseling has been a major challenge, is mostly based on theoretical data, and can be improved by the genotype–phenotype correlation studies and molecular cytogenetic analysis in which the chromosomal origins of the sSMC are detected (7). The chromosomal origin of sSMC must be detected to establish a reliable genotype–phenotype correlation (8). In our case, the prenatal molecular cytogenetic assay led to the detection of a de novo sSMCs derived from the proximal region of the long arm of chromosome 4 that resulted in trisomy of 4q11q12. The individuals carrying very small 4q11eq13proximal duplications seem healthy with normal features, however, they may have learning disability and developmental delay (4). However, the duplications of the proximal region of 4q have been contributed to different abnormalities and clinically important features. For example, a 4q12eq13 duplication in a 6-yr-old girl with microcephaly, facial dimorphism clinodactyly of the fifth finger, and psychomotor retardation has been detected (9). A 47,XY,+r [4] (::p10/q12::) karyotype in a 27-yr-old male with facial dimorphism, severe mental retardation, language disability, syndactyly of foot, as well as clinodactyly of the hand has reported (10). A 15-yr-old girl with 4q13.1eq22.2 duplication, who had minor physical anomalies and moderate intellectual disabilities has been reported (11). A 2-yr-and-8-month-old boy with duplication of 4q12eq13 who had microcephaly, mild facial dimorphism, and mental retardation has been previously reported (12). Bonnet and co-workers reported a six-yr-old obese girl with a developmental delay who had 82% mosaicism for an sSMC [4] 4q10eq13 in peripheral lymphocytes (13). An eight-yr-old girl with 8.6-Mb duplication of 4q13.1eq13.3 with developmental delay, attention-deficit hyperactivity, and speaking disability has been previously reported (14). A 47, XX, +mar has been detected with 4p11eq12 sSMC in which only long philtrum and hypertelorism were observed at the termination of pregnancy (15). A three-yr-old boy with 4p11eq12-derived sSMC [4] presenting developmental delay, mild motor retardation, and mild hypotonic features has also been shown (7). As mentioned, de novo sSMCs were not indicated in any of aforementioned studies, and such de novo sSMCs may be undetected causing major clinical manifestations. Our study provides useful information for genetic counseling on prenatally detectable sSMC of 4q11q12.
4. Conclusion
It has been concluded that if the marker chromosome is seen in the amniotic fluid sample but does not appear in parents, the CGH array is needed for making the best decision. Such findings help us in concise genetic counseling and guidance of couples making proper decisions about their fetuses.
Acknowledgements
The authors of this study thank the personnel of the genetic laboratory of Shiraz Fertility Center, Shiraz, Iran, for their kind cooperation.
Conflict of Interest
The authors have no conflicts of interest relevant to this article.
Type of Study: Case Report |
Subject:
Reproductive Genetics
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
