
International Journal of
Reproductive Biomedicine
Mon, Jul 20, 2026
[Archive]
Volume 20, Issue 12 (December 2022)
IJRM 2022, 20(12): 1047-1050 |
Back to browse issues page




![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Hassanlou M, Abiri M, Zeinali S. Prenatal diagnosis of citrullinemia type 1; seven families with c.1168G>A mutation of Argininosuccinate synthetase 1 gene in Southwest Iran: A case series. IJRM 2022; 20 (12) :1047-1050
URL: http://ijrm.ir/article-1-2318-en.html
URL: http://ijrm.ir/article-1-2318-en.html



1- Farzanegan Campus, Semnan University, Semnan, Iran. , m.hassanlou@semnan.ac.ir
2- Department of Medical Genetics, Faculty of Medicine, Iran University of Medical Sciences, Tehran, Iran. Shahid Akbarabadi Clinical Research Development Unit, Iran University of Medical Sciences, Tehran, Iran.
3- Dr. Zeinali's Medical Genetics Laboratory, Kawsar Human Genetics Research Center, Tehran, Iran. Department of Molecular Medicine, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran.
2- Department of Medical Genetics, Faculty of Medicine, Iran University of Medical Sciences, Tehran, Iran. Shahid Akbarabadi Clinical Research Development Unit, Iran University of Medical Sciences, Tehran, Iran.
3- Dr. Zeinali's Medical Genetics Laboratory, Kawsar Human Genetics Research Center, Tehran, Iran. Department of Molecular Medicine, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran.
Full-Text [PDF 644 kb]
(1694 Downloads)
| Abstract (HTML) (2363 Views)
1. Introduction
Citrullinemia type 1 is an autosomal recessive rare, life-threatening inherited disease in 1:57,000 births with higher incidences in populations with more frequent consanguinity (1). Citrullinemia type 1 includes a classic neonatal acute form, a late-onset milder form, a form that begins during or after pregnancy, and an asymptomatic form. The underlying biochemical defect is a deficiency of the argininosuccinate synthetase (ASS) enzyme due to a mutation in the ASS1 gene. ASS is one of the 6 enzymes that have a role in the urea cycle, which removes nitrogen from the body (2). The synthesis of argininosuccinate from citrulline and aspartate is catalyzed by ASS enzyme-the 3rd step in the urea cycle. The enzyme deficiency is characterized by elevated blood citrulline and ammonia levels, which often results in hyperammonemia coma and early neonatal death in affected children due to cerebral edema and encephalopathy if left untreated. In classic form of Citrullinemia several symptom including vomiting, refusal to eat, progressive lethargy, and signs of increased intracranial pressure are seen in infants. Prompt treatment is needed for prolonged survival. The main treatments for ASS deficiency is a low protein and high-calorie diet supplemented with amino acid, particularly arginine. Severe hyperammonemia could result in neurological damage and can be treated with hemodiafiltration (3, 4). An untreated infant with early-onset citrullinemia type 1 is expected to live only 17 days as the longest reported survival time (5). In a few late-onset forms of the disease, symptomatic hyperammonemia develops during childhood and adulthood (6).
The ASS1 gene has 64 kb length located in chromosome 9q34.1, with 16 exons encoding 412 amino acids. In the ASS1 gene, several mutations have been reported, of which p.Gly390Arg, p.Arg363Trp, and p.Gly14Ser are the most pathogenic variants with the early onset and severe phenotype (7). The most common mutation is p.Gly390Arg (c.1168G>A) in exon 15, in multiple ethnic backgrounds including the USA, Canada, Spain, Austria, etc. (8). The residue G390 is conserved in ASS1 gene sequences and is located in α-helix 12, which is critical for multimerization (9, 10). Only a few citrullinemia type 1 have been reported in the Iranian population (11), and none of the prenatal diagnosis (PND) was reported.
In this study, for the first time, we report the identification of Iranian patients with citrullinemia type I in a limited geographic area and postulate a possible population cluster.
2. Cases presentation
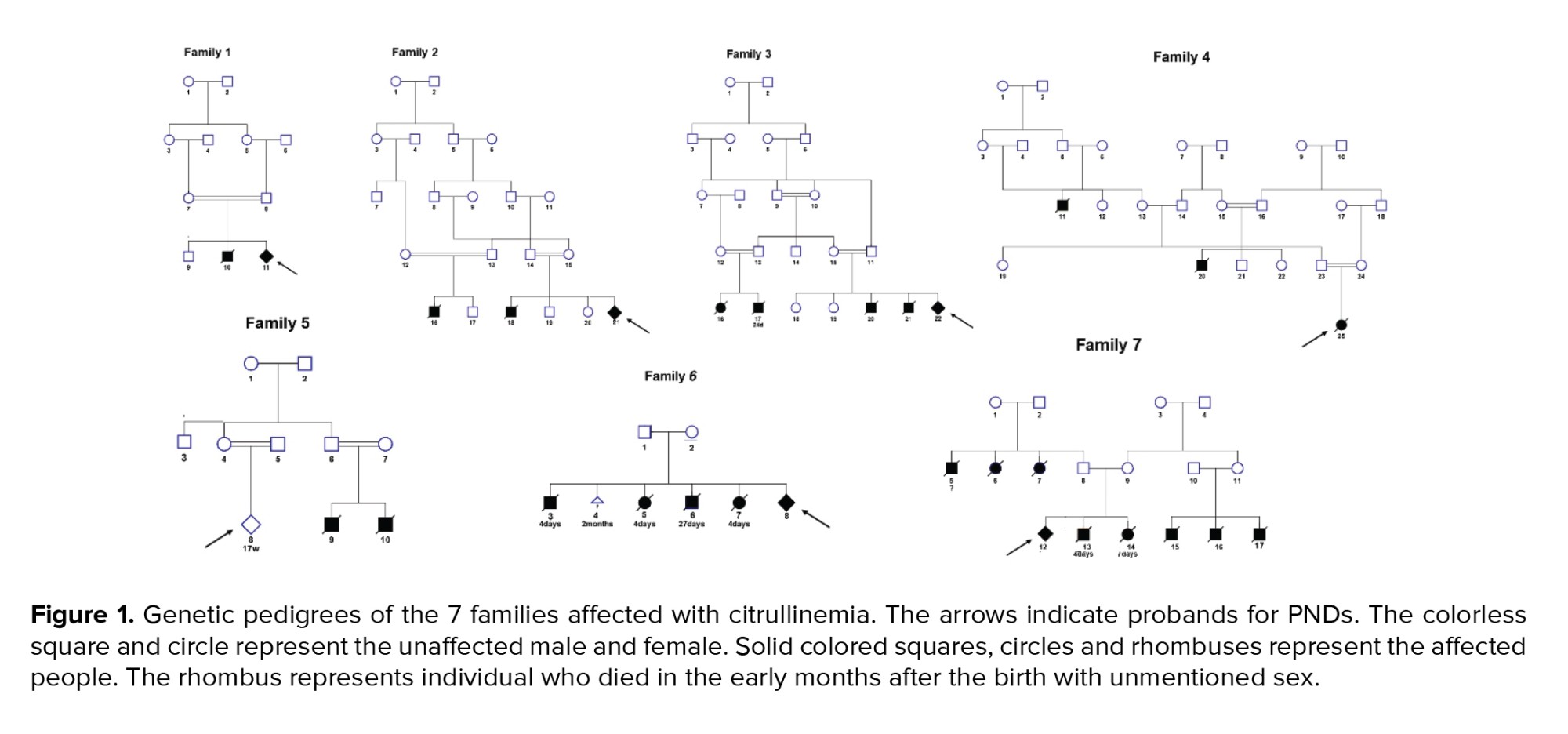
7 Iranian families from Southwest Iran from 2017-2020, having one or more children in their families and/or relatives, who died in the early months after birth due to citrullinemia type 1, were studied (Figure 1). Couples came at 12-14 wk of pregnancy for PND genetic counseling.
Sanger sequencing and haplotyping techniques were performed on couples to confirm their carrier status. To this end, 5 ml of blood was collected from each parent in a heparin / EDTA vial. For chorionic villus sampling from 11-14 wk gestation, duration of pregnancy weeks was confirmed by the gynecologist under ultrasound guidance using a transabdominal approach.
DNA extraction was done on blood and fetal samples by the Genomic DNA Purification Kit (Invitrogen, USA) according to the manufacturer’s instructions. Using Nanodrop (Invitrogen) the purity and concentration of extracted DNA were tested.
Whole-exome sequencing was done on all exomes of protein-coding genes and other important genomic regions. Next-generation sequencing was done on Illumina Sequencer to sequence about 100 million reads. Sequencing results were analyzed using international bioinformatics databases and standard software.
For haplotyping, single-nucleotide polymorphisms were selected for areas flanking the ASS1 gene (dbSNP, build 151). PCR primers were designed with Primer3. The short tandem repeat typing method was done by gel electrophoresis.
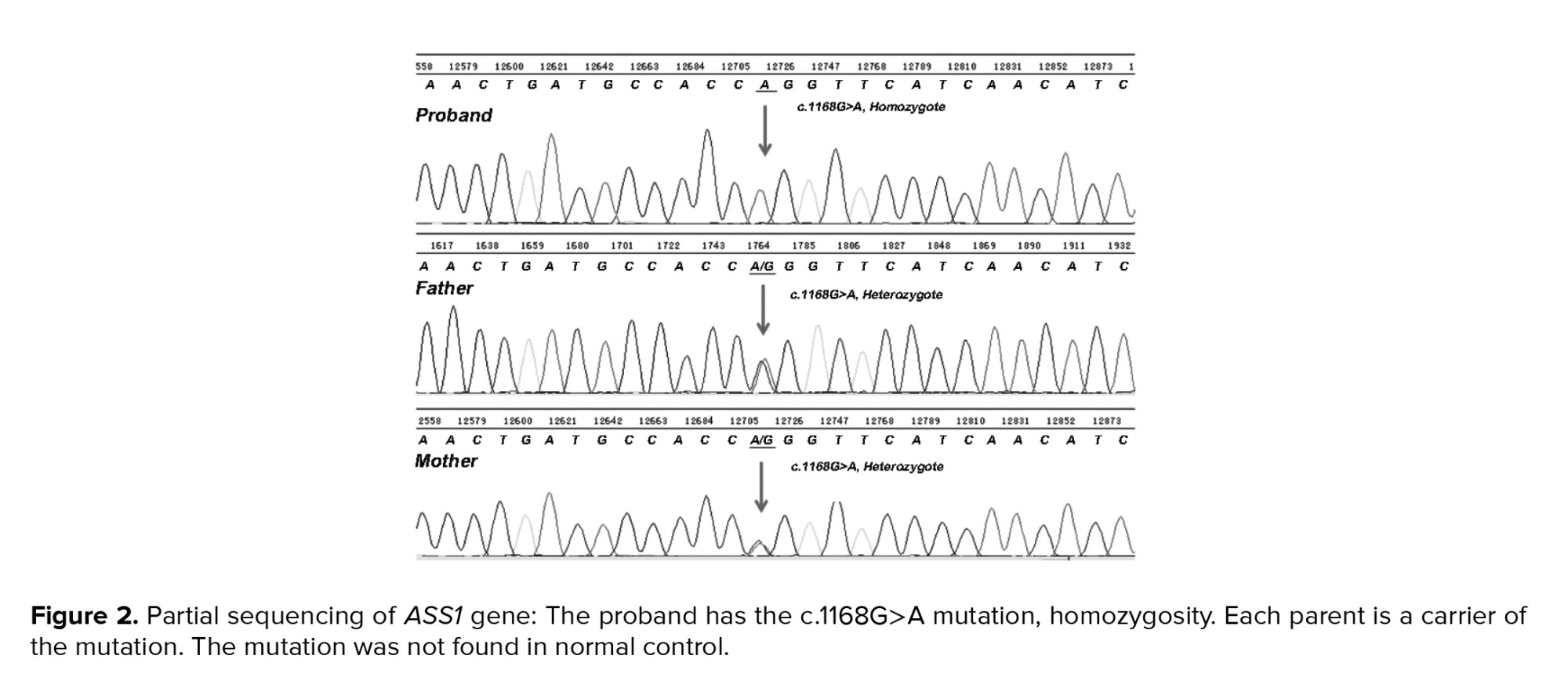
According to the whole-exome sequencing analysis and haplotyping, all the families except for the 5th family had a fetus with homozygote missense mutation for ASS1 in c.1165G>A DNA sequences, which resulted in Gly to Lys substitution (Figure 2).
2.1. Ethical considerations
Ethical approval was obtained from Kowsar Human Genetics Research Center Ethics Committee, Tehran, Iran with the number of 14006319. Written informed consent form was obtained from the child's parents.
3. Discussion
In this study, the PND of citrullinemia type 1 was performed in 7 Iranian families from Southwest Iran having one or more children or their relatives children who died in the early months after the birth. Whole-exome sequencing of the coding exons was performed in the parents of the index cases. Haplotype mapping indicated that they were heterozygous for the p.G390R substitution; this mutation was then searched in the fetus. The homozygous substitution was detected in the proband of 6 among 7 families.
The p.Gly390Arg substitution is the most common mutation located in a highly conserved region of exon 15 of the ASS1 gene. This missense mutation is located in the protein helix and results in the enzymatic inactivation, demonstrating the vitality of this region for multimerization and proper enzyme functioning (12). The homozygous state exclusively results in severely affected patients (13). Other mutations have been found in the ASS1 gene, but the rate of occurrence is rare (14).
The p.G390R substitution mutation in the ASS1 gene is a recessive lethal mutation, which means that the inheritance of 2 recessive lethal alleles is fatal. Cystic fibrosis, sickle-cell anemia, and achondroplasia are other examples of human diseases caused by the recessive lethal alleles (15). According to Mendelian inheritance of monogenic disease, following mating between 2 heterozygotes or carriers, 25% of offspring may be recessive for 2 alleles (16-18). In our study, families 6 and 7 had a large number of affected infants. Because citrullinemia type 1 is an autosomal recessive disease and is likely to be inherited from carrier parents by 25% probability in each pregnancy, with current information about this disease, the large number of affected infants in families 6 and 7 is accidental. Further studies are needed to better understand the inheritance behavior of the ASS1 gene (19).
Our study identified a p.Gly390Arg mutation in ASS1, which may be shared among individuals of Southwest Iran and demonstrate a possible population cluster. This information is crucial to identify the carriers in genetic counseling. In addition, this knowledge could increase clinician's awareness in the diagnosis of new cases in a very early neonatal period.
4. Conclusion
p.Gly390Arg mutation in ASS1 shared among individuals of Southwest Iran which demonstrate a possible population cluster.
Acknowledgments
This research has not received any specific grant from any funding sectors. This work was supported by the Kawsar Human Genetics Research Center, Tehran, Iran.
Conflict of Interest
The authors have no conflict of interest.
Full-Text: (398 Views)
1. Introduction
Citrullinemia type 1 is an autosomal recessive rare, life-threatening inherited disease in 1:57,000 births with higher incidences in populations with more frequent consanguinity (1). Citrullinemia type 1 includes a classic neonatal acute form, a late-onset milder form, a form that begins during or after pregnancy, and an asymptomatic form. The underlying biochemical defect is a deficiency of the argininosuccinate synthetase (ASS) enzyme due to a mutation in the ASS1 gene. ASS is one of the 6 enzymes that have a role in the urea cycle, which removes nitrogen from the body (2). The synthesis of argininosuccinate from citrulline and aspartate is catalyzed by ASS enzyme-the 3rd step in the urea cycle. The enzyme deficiency is characterized by elevated blood citrulline and ammonia levels, which often results in hyperammonemia coma and early neonatal death in affected children due to cerebral edema and encephalopathy if left untreated. In classic form of Citrullinemia several symptom including vomiting, refusal to eat, progressive lethargy, and signs of increased intracranial pressure are seen in infants. Prompt treatment is needed for prolonged survival. The main treatments for ASS deficiency is a low protein and high-calorie diet supplemented with amino acid, particularly arginine. Severe hyperammonemia could result in neurological damage and can be treated with hemodiafiltration (3, 4). An untreated infant with early-onset citrullinemia type 1 is expected to live only 17 days as the longest reported survival time (5). In a few late-onset forms of the disease, symptomatic hyperammonemia develops during childhood and adulthood (6).
The ASS1 gene has 64 kb length located in chromosome 9q34.1, with 16 exons encoding 412 amino acids. In the ASS1 gene, several mutations have been reported, of which p.Gly390Arg, p.Arg363Trp, and p.Gly14Ser are the most pathogenic variants with the early onset and severe phenotype (7). The most common mutation is p.Gly390Arg (c.1168G>A) in exon 15, in multiple ethnic backgrounds including the USA, Canada, Spain, Austria, etc. (8). The residue G390 is conserved in ASS1 gene sequences and is located in α-helix 12, which is critical for multimerization (9, 10). Only a few citrullinemia type 1 have been reported in the Iranian population (11), and none of the prenatal diagnosis (PND) was reported.
In this study, for the first time, we report the identification of Iranian patients with citrullinemia type I in a limited geographic area and postulate a possible population cluster.
2. Cases presentation
7 Iranian families from Southwest Iran from 2017-2020, having one or more children in their families and/or relatives, who died in the early months after birth due to citrullinemia type 1, were studied (Figure 1). Couples came at 12-14 wk of pregnancy for PND genetic counseling.
Sanger sequencing and haplotyping techniques were performed on couples to confirm their carrier status. To this end, 5 ml of blood was collected from each parent in a heparin / EDTA vial. For chorionic villus sampling from 11-14 wk gestation, duration of pregnancy weeks was confirmed by the gynecologist under ultrasound guidance using a transabdominal approach.
DNA extraction was done on blood and fetal samples by the Genomic DNA Purification Kit (Invitrogen, USA) according to the manufacturer’s instructions. Using Nanodrop (Invitrogen) the purity and concentration of extracted DNA were tested.
Whole-exome sequencing was done on all exomes of protein-coding genes and other important genomic regions. Next-generation sequencing was done on Illumina Sequencer to sequence about 100 million reads. Sequencing results were analyzed using international bioinformatics databases and standard software.
For haplotyping, single-nucleotide polymorphisms were selected for areas flanking the ASS1 gene (dbSNP, build 151). PCR primers were designed with Primer3. The short tandem repeat typing method was done by gel electrophoresis.
According to the whole-exome sequencing analysis and haplotyping, all the families except for the 5th family had a fetus with homozygote missense mutation for ASS1 in c.1165G>A DNA sequences, which resulted in Gly to Lys substitution (Figure 2).
2.1. Ethical considerations
Ethical approval was obtained from Kowsar Human Genetics Research Center Ethics Committee, Tehran, Iran with the number of 14006319. Written informed consent form was obtained from the child's parents.
3. Discussion
In this study, the PND of citrullinemia type 1 was performed in 7 Iranian families from Southwest Iran having one or more children or their relatives children who died in the early months after the birth. Whole-exome sequencing of the coding exons was performed in the parents of the index cases. Haplotype mapping indicated that they were heterozygous for the p.G390R substitution; this mutation was then searched in the fetus. The homozygous substitution was detected in the proband of 6 among 7 families.
The p.Gly390Arg substitution is the most common mutation located in a highly conserved region of exon 15 of the ASS1 gene. This missense mutation is located in the protein helix and results in the enzymatic inactivation, demonstrating the vitality of this region for multimerization and proper enzyme functioning (12). The homozygous state exclusively results in severely affected patients (13). Other mutations have been found in the ASS1 gene, but the rate of occurrence is rare (14).
The p.G390R substitution mutation in the ASS1 gene is a recessive lethal mutation, which means that the inheritance of 2 recessive lethal alleles is fatal. Cystic fibrosis, sickle-cell anemia, and achondroplasia are other examples of human diseases caused by the recessive lethal alleles (15). According to Mendelian inheritance of monogenic disease, following mating between 2 heterozygotes or carriers, 25% of offspring may be recessive for 2 alleles (16-18). In our study, families 6 and 7 had a large number of affected infants. Because citrullinemia type 1 is an autosomal recessive disease and is likely to be inherited from carrier parents by 25% probability in each pregnancy, with current information about this disease, the large number of affected infants in families 6 and 7 is accidental. Further studies are needed to better understand the inheritance behavior of the ASS1 gene (19).
Our study identified a p.Gly390Arg mutation in ASS1, which may be shared among individuals of Southwest Iran and demonstrate a possible population cluster. This information is crucial to identify the carriers in genetic counseling. In addition, this knowledge could increase clinician's awareness in the diagnosis of new cases in a very early neonatal period.
4. Conclusion
p.Gly390Arg mutation in ASS1 shared among individuals of Southwest Iran which demonstrate a possible population cluster.
Acknowledgments
This research has not received any specific grant from any funding sectors. This work was supported by the Kawsar Human Genetics Research Center, Tehran, Iran.
Conflict of Interest
The authors have no conflict of interest.
Type of Study: Case Report |
Subject:
Reproductive Genetics
References
1. Nuciforo S, Heim MH. Organoids to model liver disease. JHEP Rep 2020; 3: 100198. [DOI:10.1016/j.jhepr.2020.100198] [PMID] [PMCID]
2. Zielonka M, Kölker S, Gleich F, Stützenberger N, Nagamani SCS, Gropman AL, et al. Early prediction of phenotypic severity in citrullinemia type 1. Ann Clin Transl Neurol 2019; 6: 1858-1871. [DOI:10.1002/acn3.50886] [PMID] [PMCID]
3. Kang H, Kim M, Lee JH. Nutritional management in a patient with citrullinemia type 1. Clin Nutr Res 2021; 10: 268-277. [DOI:10.7762/cnr.2021.10.3.268] [PMID] [PMCID]
4. Hayasaka K. Metabolic basis and treatment of citrin deficiency. J Inherit Metab Dis 2021; 44: 110-117. [DOI:10.1002/jimd.12294] [PMID]
5. Szlosarek PW, Steele JP, Nolan L, Gilligan D, Taylor P, Spicer J, et al. Arginine deprivation with pegylated arginine deiminase in patients with argininosuccinate synthetase 1-deficient malignant pleural mesothelioma: A randomized clinical trial. JAMA Oncol 2017; 3: 58-66. [DOI:10.1001/jamaoncol.2016.3049] [PMID]
6. Ruxmohan S, Quinonez J, Choudhari J, Poudel S, Pandav K. Hyperammonemic encephalopathy in an adolescent patient of citrullinemia type 1 with an atypical presentation. Cureus 2021; 13: e15109. [DOI:10.7759/cureus.15109]
7. Lin Y, Gao H, Lu B, Zhou S, Zheng T, Lin W, et al. Citrullinemia type I is associated with a novel splicing variant, c.773 + 4A > C, in ASS1: A case report and literature review. BMC Med Genet 2019; 20: 110. [DOI:10.1186/s12881-019-0836-5] [PMID] [PMCID]
8. Randon DN, Sperb-Ludwig F, Vianna FSL, Becker APP, Vargas CR, Sitta A, et al. Prevalence of the most common pathogenic variants in three genes for inborn errors of metabolism associated with sudden unexpected death in infancy: A population-based study in south Brazil. Genet Mol Biol 2020; 43: 20190298. [DOI:10.1590/1678-4685-gmb-2019-0298] [PMID] [PMCID]
9. Diez-Fernandez C, Rüfenacht V, Häberle J. Mutations in the human argininosuccinate synthetase (ASS1) gene, impact on patients, common changes, and structural considerations. Hum Mutat 2017; 38: 471-484. [DOI:10.1002/humu.23184] [PMID]
10. Diez-Fernandez C, Wellauer O, Gemperle C, Rüfenacht V, Fingerhut R, Häberle J. Kinetic mutations in argininosuccinate synthetase deficiency: Characterisation and in vitro correction by substrate supplementation. J Med Genet 2016; 53: 710-719. [DOI:10.1136/jmedgenet-2016-103937] [PMID]
11. Pishva N, Mirzaee A, Karamizade Z, Pourarian S, Hemmati F, Razvi M, et al. Selective screening of 650 high risk Iranian patients for detection of inborn error of metabolism. Iran J Neonat 2015; 5: 11-14.
12. Diez-Fernandez C, Rüfenacht V, Häberle J. Mutations in the human argininosuccinate synthetase (ASS1) gene, impact on patients, common changes, and structural considerations. Hum Mutat 2017; 38: 471-484. [DOI:10.1002/humu.23184] [PMID]
13. Moarefian Sh, Zamani M, Rahmanifar A, Behnam B, Zaman T. Clinical, laboratory data and outcomes of 17 Iranian citrullinemia type 1 patients: Identification of five novel ASS1 gene mutations. JIMD Rep 2022; 63: 231-239. [DOI:10.1002/jmd2.12277] [PMID] [PMCID]
14. Silvera-Ruiz SM, Arranz JA, Häberle J, Angaroni CJ, Bezard M, Guelbert N, et al. Urea cycle disorders in argentine patients: Clinical presentation, biochemical and genetic findings. Orphanet J Rare Dis 2019; 14: 203. [DOI:10.1186/s13023-019-1177-3] [PMID] [PMCID]
15. Kose E, Unal O, Bulbul S, Gunduz M, Häberle J, Arslan N. Identification of three novel mutations in fourteen patients with citrullinemia type 1. Clin Biochem 2017; 50: 686-689. [DOI:10.1016/j.clinbiochem.2017.01.011] [PMID]
16. Castellani C, Assael BM. Cystic fibrosis: A clinical view. Cell Mol Life Sci 2017; 74: 129-140. [DOI:10.1007/s00018-016-2393-9] [PMID]
17. Venugopal A, Chandran M, Eruppakotte N, Kizhakkillach S, Breezevilla SC, Vellingiri B. Monogenic diseases in India. Mutat Res Rev Mutat Res 2018; 776: 23-31. [DOI:10.1016/j.mrrev.2018.03.003] [PMID]
18. Kausar M, Mäkitie RE, Toiviainen-Salo S, Ignatius J, Anees M, Mäkitie O. Recessive multiple epiphyseal dysplasia- Clinical characteristics caused by rare compound heterozygous SLC26A2 genotypes. Eur J Med Genet 2019; 62: 103573. [DOI:10.1016/j.ejmg.2018.11.007] [PMID]
19. Chakravarti A. Magnitude of mendelian versus complex inheritance of rare disorders. Am J Med Genet A 2021; 185: 3287-3293. [DOI:10.1002/ajmg.a.62463] [PMID]
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
