
International Journal of
Reproductive Biomedicine
Tue, Aug 4, 2026
[Archive]
Volume 23, Issue 6 (June 2025)
IJRM 2025, 23(6): 517-526 |
Back to browse issues page



Ethics code: IR.IAU.VARAMIN.REC.1401.006
![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Ashrafzadeh H, Tafvizi F, Ghasemi N, Vahidi Mehrjardi M Y, Naseh V. Prenatal diagnosis using next-generation sequencing in genetic counseling: Novel mutations in three large Iranian families: A case series. IJRM 2025; 23 (6) :517-526
URL: http://ijrm.ir/article-1-3576-en.html
URL: http://ijrm.ir/article-1-3576-en.html
Hamidreza Ashrafzadeh1 

 , Farzaneh Tafvizi1 , Nasrin Ghasemi2 , Mohammad Yahya Vahidi Mehrjardi3 , Vahid Naseh *4
, Farzaneh Tafvizi1 , Nasrin Ghasemi2 , Mohammad Yahya Vahidi Mehrjardi3 , Vahid Naseh *4
, Farzaneh Tafvizi1 , Nasrin Ghasemi2 , Mohammad Yahya Vahidi Mehrjardi3 , Vahid Naseh *4
1- Department of Biology, Pa.c., Islamic Azad University, Parand, Iran.
2- Abortion Research Center, Yazd Reproductive Sciences Institute, Shahid Sadoughi University of Medical Sciences, Yazd, Iran.
3- Diabetes Research Center, Shahid Sadoughi University of Medical Sciences, Yazd, Iran.
4- Department of Biology, Pa.c., Islamic Azad University, Parand, Iran. ,vahid55vet@yahoo.com
2- Abortion Research Center, Yazd Reproductive Sciences Institute, Shahid Sadoughi University of Medical Sciences, Yazd, Iran.
3- Diabetes Research Center, Shahid Sadoughi University of Medical Sciences, Yazd, Iran.
4- Department of Biology, Pa.c., Islamic Azad University, Parand, Iran. ,
Keywords: Prenatal diagnosis, Genetic counselling, Whole exome sequencing, Intellectual disability, Developmental delay.
Full-Text [PDF 561 kb]
(561 Downloads)
| Abstract (HTML) (890 Views)
Full-Text: (129 Views)
1. Introduction
Genetic diseases arise from various causes, notably chromosomal and single-gene disorders. Their investigation involves multiple methods to identify underlying causes. Pedigree analysis clarifies inheritance patterns and the role of chromosomal or single-gene abnormalities. For cases without chromosomal defects, molecular testing is key. Due to the genetic heterogeneity of many conditions, whole exome sequencing (WES) is among the most effective diagnostic tools (1).
Common genetically heterogeneous disorders such as developmental delay (DD), hearing loss, epilepsy, and cardiomyopathy can significantly affect individuals and their families, making genetic counseling essential. For at-risk families, prenatal diagnosis (PND) enables assessment of these conditions in the fetus, supporting informed decisions about pregnancy management and future reproductive options. Among these, intellectual disability (ID) imposes a considerable health burden and has a well-established genetic basis. Approximately 5% of ID cases are linked to cytogenetic alterations, such as aneuploidies, detectable via karyotyping, while 14% are attributed to copy number variants (CNVs) identified by array comparative genomic hybridization (CGH) (2, 3). Furthermore, mutations in numerous autosomal recessive and X-linked genes contribute to ID. Recent advances using next-generation sequencing (NGS) have revealed the role of de novo mutations in these disorders (4).
This study applies WES to 3 large Iranian families with ID/DD referees to Yazd Reproductive Sciences Institute, Yazd, Iran. These findings emphasize the critical role of comprehensive genetic screening, including the potential for PND in at-risk pregnancies, to improve our understanding of the genetic basis of ID and other common genetic diseases.
The field of medical genomics has undergone a revolution thanks to NGS, which makes it possible to generate large amounts of high-throughput DNA sequence data at a much lower cost than Sanger sequencing. PND through advanced technologies like NGS has emerged as a game-changer, enabling the early detection of de novo mutations and rare variants that traditional methods such as karyotyping (detecting ~5% of cases) or array CGH (~14% via CNVs) overlook. Early identification of these genetic anomalies prenatally can pave the way for therapeutic interventions, genetic counseling, and informed reproductive decisions, potentially alleviating the lifelong impact of mental retardation (4).
It is well known that ID has a genetic basis. Approximately 5% of cases are caused by cytogenetic alterations, which include aneuploidies, duplications, deletions, translocations, and inversions, detectable by routine karyotyping. Additionally, 14% of cases are explained by lower CNVs found using array CGH.
These disorders are known to be caused by mutations in more than 50 autosomal recessive genes and more than 100 genes of the X chromosome. The importance of de novo mutations in the etiology of these disorders has become apparent in recent years largely due to the increasing use of NGS (5, 6). This study aimed to explore the power of WES in uncovering genetic variants in couples who are seeking genetic counselling for their next pregnancy.
2. Case Presentations
This study examines 5 cases from 3 prominent Iranian families, providing valuable insights into the genetic diversity and mutation prevalence within the Iranian population. Each family included at least one child diagnosed with a developmental and/or ID, which was the primary reason for their participation in this study. Participants were referred to the genetic counseling clinic at the Abortion Research Center and Yazd Reproductive Science Institute, Yazd, Iran, aiming for favorable outcomes in subsequent pregnancies. The 5 selected samples were first subjected to cytogenetic analysis to check for chromosomal abnormalities. We found no evidence of karyotype changes in the affected individuals. Normal karyotypes confirmed WES’s necessity for detecting single-gene variants.
Unexplained ID refers to individuals who despite extensive prior evaluations, including karyotype analysis, assessments of inherited metabolic disorders, and family history could not achieve an etiological diagnosis. The inclusion criteria for this study were as follows:
Genetic diseases arise from various causes, notably chromosomal and single-gene disorders. Their investigation involves multiple methods to identify underlying causes. Pedigree analysis clarifies inheritance patterns and the role of chromosomal or single-gene abnormalities. For cases without chromosomal defects, molecular testing is key. Due to the genetic heterogeneity of many conditions, whole exome sequencing (WES) is among the most effective diagnostic tools (1).
Common genetically heterogeneous disorders such as developmental delay (DD), hearing loss, epilepsy, and cardiomyopathy can significantly affect individuals and their families, making genetic counseling essential. For at-risk families, prenatal diagnosis (PND) enables assessment of these conditions in the fetus, supporting informed decisions about pregnancy management and future reproductive options. Among these, intellectual disability (ID) imposes a considerable health burden and has a well-established genetic basis. Approximately 5% of ID cases are linked to cytogenetic alterations, such as aneuploidies, detectable via karyotyping, while 14% are attributed to copy number variants (CNVs) identified by array comparative genomic hybridization (CGH) (2, 3). Furthermore, mutations in numerous autosomal recessive and X-linked genes contribute to ID. Recent advances using next-generation sequencing (NGS) have revealed the role of de novo mutations in these disorders (4).
This study applies WES to 3 large Iranian families with ID/DD referees to Yazd Reproductive Sciences Institute, Yazd, Iran. These findings emphasize the critical role of comprehensive genetic screening, including the potential for PND in at-risk pregnancies, to improve our understanding of the genetic basis of ID and other common genetic diseases.
The field of medical genomics has undergone a revolution thanks to NGS, which makes it possible to generate large amounts of high-throughput DNA sequence data at a much lower cost than Sanger sequencing. PND through advanced technologies like NGS has emerged as a game-changer, enabling the early detection of de novo mutations and rare variants that traditional methods such as karyotyping (detecting ~5% of cases) or array CGH (~14% via CNVs) overlook. Early identification of these genetic anomalies prenatally can pave the way for therapeutic interventions, genetic counseling, and informed reproductive decisions, potentially alleviating the lifelong impact of mental retardation (4).
It is well known that ID has a genetic basis. Approximately 5% of cases are caused by cytogenetic alterations, which include aneuploidies, duplications, deletions, translocations, and inversions, detectable by routine karyotyping. Additionally, 14% of cases are explained by lower CNVs found using array CGH.
These disorders are known to be caused by mutations in more than 50 autosomal recessive genes and more than 100 genes of the X chromosome. The importance of de novo mutations in the etiology of these disorders has become apparent in recent years largely due to the increasing use of NGS (5, 6). This study aimed to explore the power of WES in uncovering genetic variants in couples who are seeking genetic counselling for their next pregnancy.
2. Case Presentations
This study examines 5 cases from 3 prominent Iranian families, providing valuable insights into the genetic diversity and mutation prevalence within the Iranian population. Each family included at least one child diagnosed with a developmental and/or ID, which was the primary reason for their participation in this study. Participants were referred to the genetic counseling clinic at the Abortion Research Center and Yazd Reproductive Science Institute, Yazd, Iran, aiming for favorable outcomes in subsequent pregnancies. The 5 selected samples were first subjected to cytogenetic analysis to check for chromosomal abnormalities. We found no evidence of karyotype changes in the affected individuals. Normal karyotypes confirmed WES’s necessity for detecting single-gene variants.
Unexplained ID refers to individuals who despite extensive prior evaluations, including karyotype analysis, assessments of inherited metabolic disorders, and family history could not achieve an etiological diagnosis. The inclusion criteria for this study were as follows:
- ID, defined as an IQ of < 70 based on the Wechsler Intelligence Scale for children.
- DD identified as a developmental quotient of < 76 on the Gesell developmental scale across 2 or more developmental areas.
- The absence of known cases of postnatal hypoxia, head injuries, poisoning, perinatal brain injuries, or infections of the central nervous system.
Pediatric neurologists evaluated each case at Shahid Sadoughi University of Medical Sciences, Yazd, Iran.
Genomic DNA was extracted from peripheral blood (AddPrep Genomic DNA Extraction Kit; Korea), and its concentration and purity were measured using Thermo Scientific's NanoDrop 8000 spectrophotometer and Invitrogen's Qubit 2.0 fluorometer. Blood samples underwent genomic DNA isolation and cytogenetic procedures following standard protocols. Peripheral blood lymphocytes were cultured, and chromosome staining was performed using G-banding as per Seabright’s methodology (1971).
Both Splice Site Prediction and Human Splicing Finder tools were employed to assess the impact of splice variants on transcripts. In line with American College of Medical Genetics and Genomics guidelines, variants were categorized as "pathogenic", "probably pathogenic", "uncertain significance", "probably benign", or "benign". Sanger sequencing was performed on index cases and other family members to confirm the identified variants.
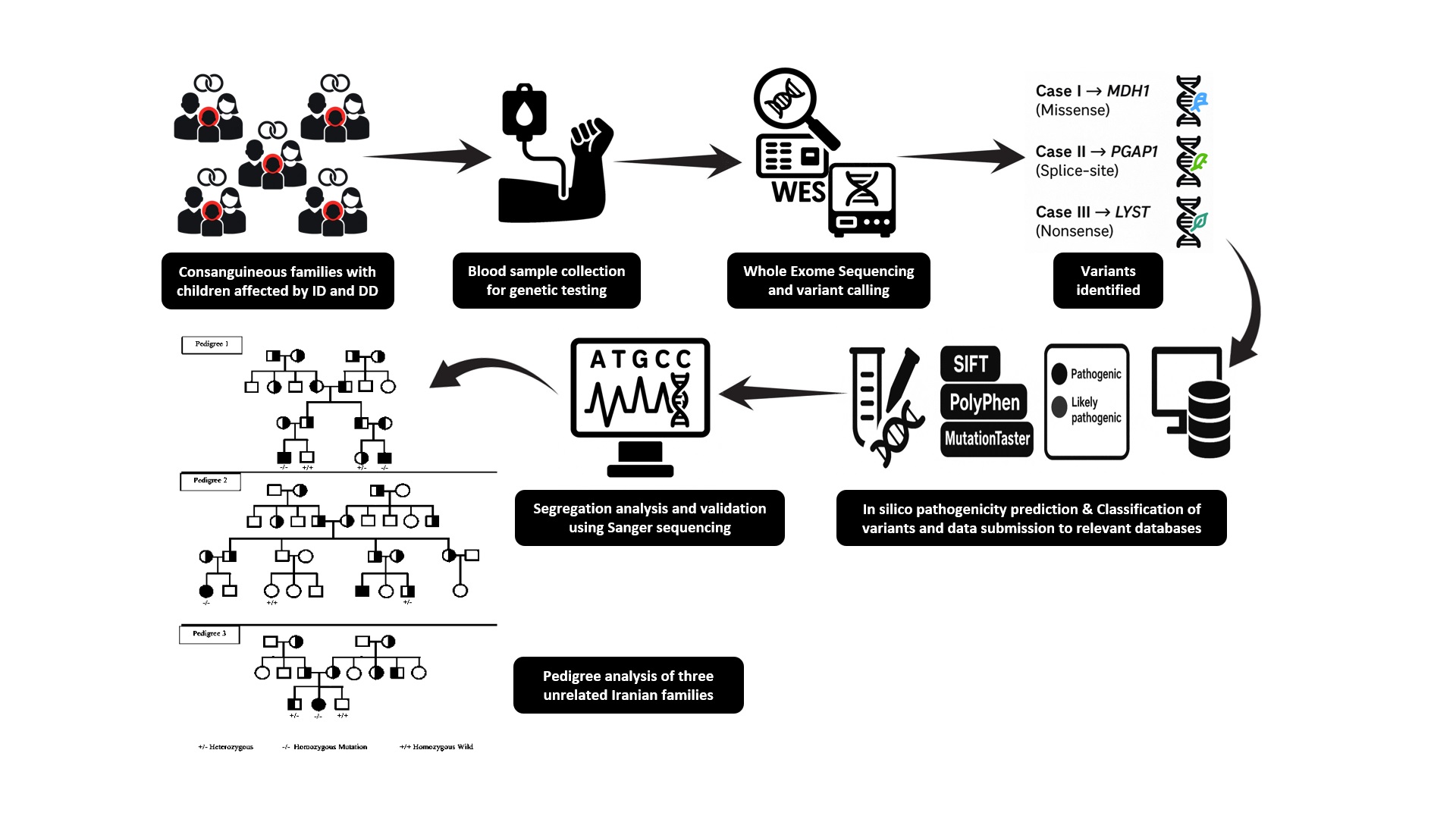
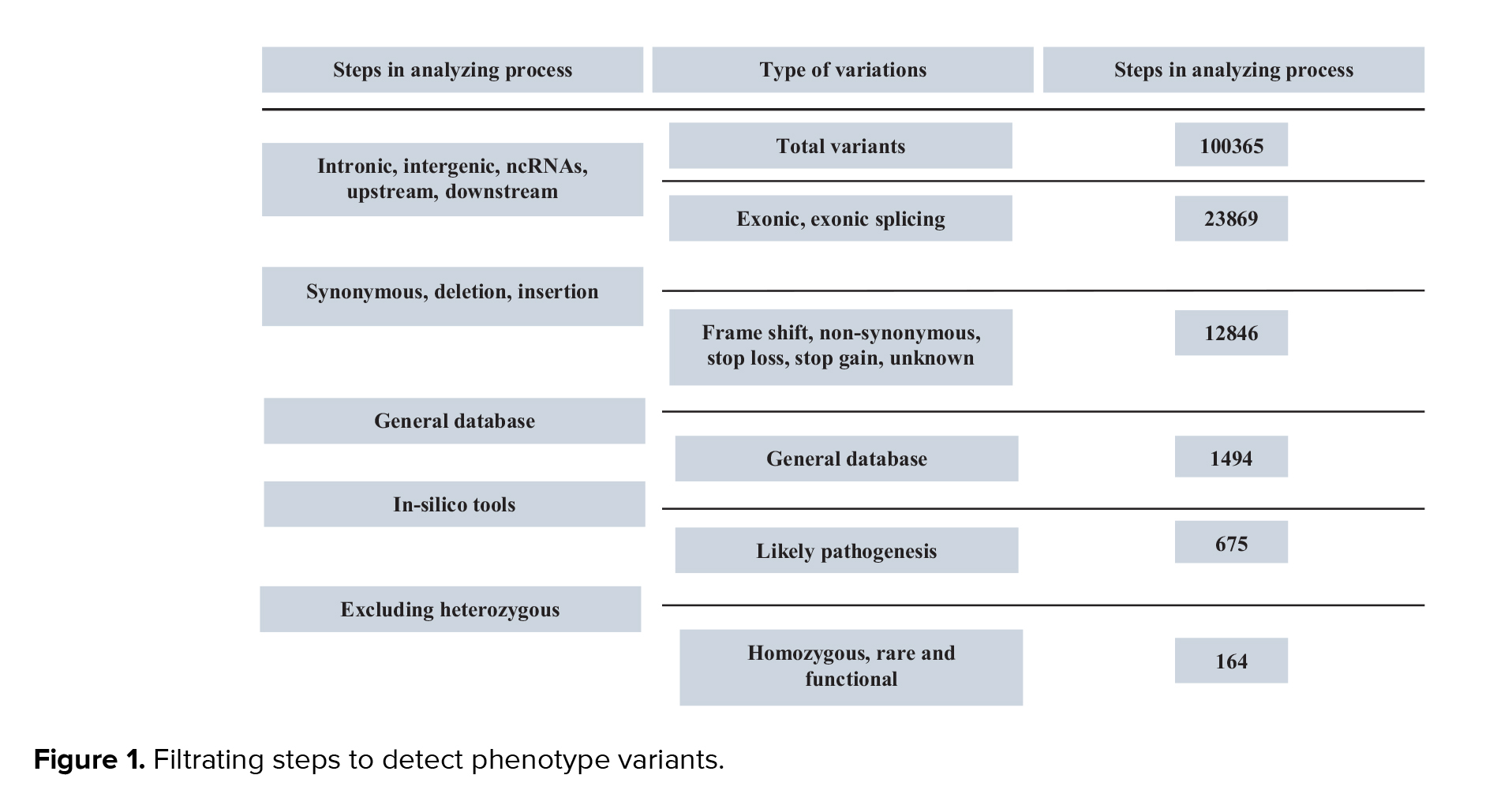
All cases, including 5 cases from 3 families, underwent WES. Variants were annotated using the variant effect predictor and filtered based on the following criteria: 1) nonsynonymous single nucleotide variants or indels located in exon or splice regions; 2) absent in control populations or with an allele frequency < 0.01 in the exome sequencing project, 1000G, ExAC or genome aggregation database (gnomAD); 3) likely to be harmful from at least 4 of the following 6 software tools: SIFT, PolyPhen-2, MutationTaster (7), CADD (8), M-CAP (9), and Condel and PROVEAN. Splice site prediction along with human splicing finder were used to estimate the impact of splice variants on transcripts. In accordance with American College of Medical Genetics and Genomics guidelines and pertinent annotation files, variants were categorized as "pathogenic", "probably pathogenic", "uncertain significance", "probably benign", or "benign". To verify the validity and distinguish between potential variants, Sanger sequencing was carried out on the index participants as well as additional family members (Figure 1).
Using the following standards, the identified variants' clinical impact was assessed: 1) a frequency of alleles < 0.03 in the 1000G or exome variant server, 2) splicing, stop-gain, and frameshift variants were all considered to be highly pathogenic, 3) for missense mutations: assessments comprised Grantham scores, SIFT, PolyPhen-2, and amino acid conservation for pathogenicity predictions, 4) various patterns of inheritance, such as dominant inheritance, recessive inheritance, or maternal inheritance of X-linked variants, 5) variant absence in other dataset (in-house database), 6) evaluate the possible influence of synonymous or intronic variants the human splicing finder web tool was utilized. Furthermore, the pathogenicity of de novo variant was further demonstrated by the use of conservation restriction statistics for every gene from the ExAC dataset (http://exac.broadinstitute.org) in conjunction with the CADD score.
Sanger sequencing verified relevant genetic variants. After the cases, his parents, and a few other family members' DNA were amplified by polymerase chain reaction using particular primers, bidirectional sequencing was carried out on an Applied Biosystem (ABI PRISM 3500) automated sequencer (life technologies) using the BigDye Terminator kit v1.1. Exon primer was used to create all the primers needed for amplification and sequencing. On request, comprehensive primer sequences and polymerase chain reactionconditions can be obtained (Table I).
2.1. Cases 1 and 2
2.1.1. Phenotype of the first family’s cases
The first family from Zahedan, Iran, comprised cases 1 and 2. Case 1 weighed 3050 gr at birth. At the age of 6 months, this participant was referred to the clinical genetics department to measure disproportionate growth parameters and growth delay.
Cranial ultrasound results were normal. At the age of 2, he weighed 14 kg and was 79 cm tall. He was classified as moderately delayed in all aspects of development, could sit independently at 11 months, and started walking between 2 and 3 yr of age. He started talking at about 6 yr old and could say simple words and was also able to follow one-step commands. This boy was sensitive to loud noises and crowded places. In the examination, at the age of 6 yr and 10 months, his height was 100 cm, and his weight reached 21 kg, which indicated relative macrocephaly. He had intermittent strabismus but was visually quite satisfactory. Unlike his fingernails, which were normal, all his toenails, especially the 4th and 5th toenails, were hypoplastic. The results of the MRI scan of the skull at the age of 9 months showed a corpus callosum with mild deformity, which delayed myelination.
Case no. 2 was referred to the clinical genetics center due to his growth and development.
He weighed 2.65 kg at birth. He had laryngomalacia and had problems such as feeding difficulty, vomiting, constipation, and sleep disturbance. The results of the developmental evaluation showed that he has a moderate DD (he started to walk at 22 months and started his first words at 20 months). This boy was short (91.8 cm) with a normal head circumference and had facial deformities, including a thin upper lip and micrognathia.
2.1.2. Genotype
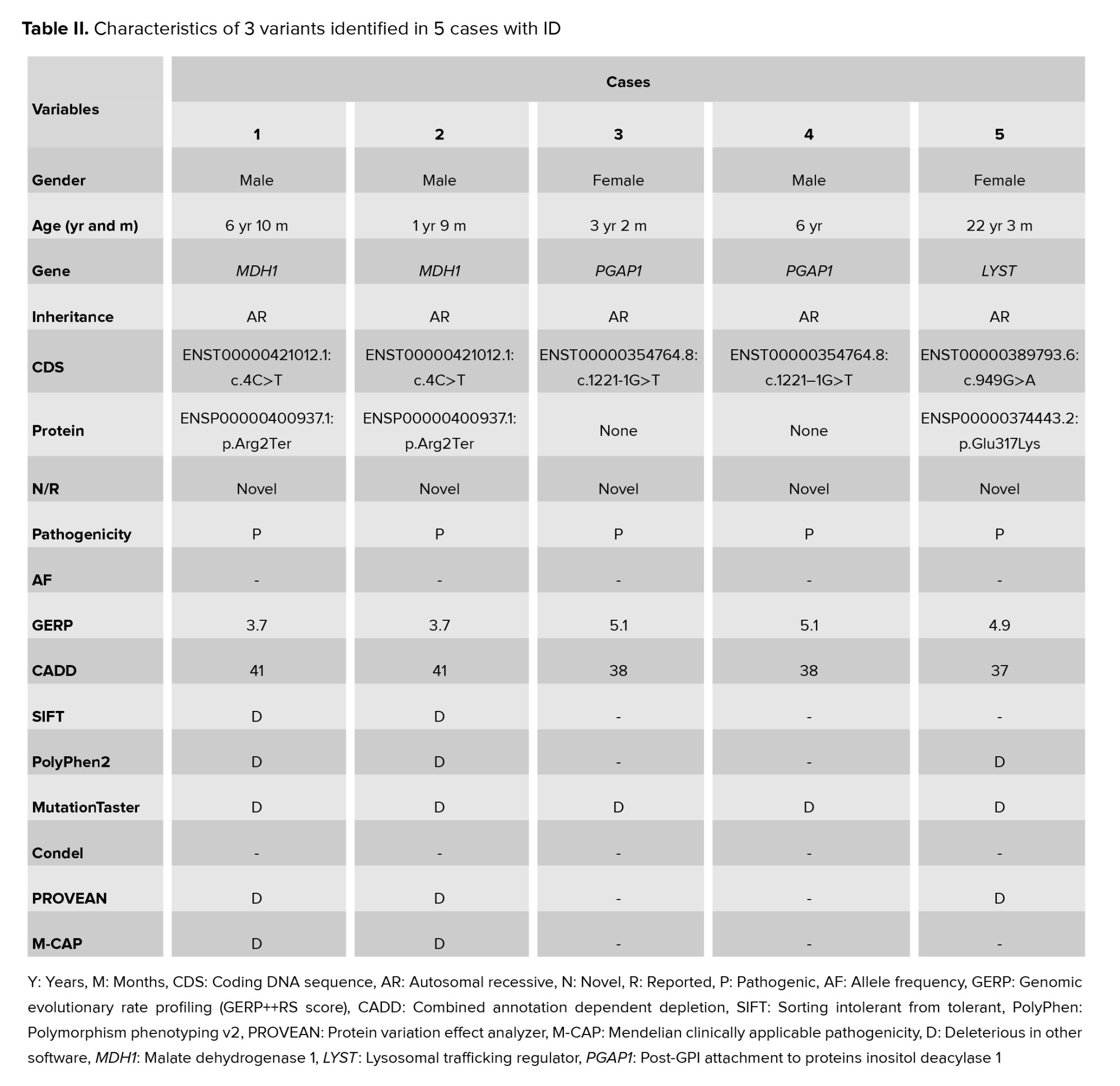
In the malate dehydrogenase 1 (MDH1) gene, a homozygous de novo stop-gain variant was found, specifically (NM_005917.4:c.4C>T). The truncation of the MDH1 protein occurs due to the introduction of an early stop codon caused by the mentioned base change.
2.2. Cases 3 and 4
2.2.1. Phenotype of the second family’s cases
Cases number 3 and 4 were from Yazd, Iran. Fetal scans revealed renal pelvic enlargement in case 3. She weighed 3100 gr at birth. She had severe feeding issues and DDS in her early years; at 14 wk, she smiled, at 13 months, she could sit by herself, and at 23 months, she could walk by herself. She was observed to be utilizing single words at the age of 2, but her primary mode of communication was nonverbal. She weighed 11.2 kg and was 87 cm tall. She experienced frequent infections and constipation as a baby. Myotonic dystrophy was ruled out, and a clinical examination revealed generalized hypotension, joint hypermobility, and slightly elevated creatine kinase levels. On assessment, she also had plagiocephaly, prominent ears, a thin upper lip of vermilion, and downturned mouth corners.
At 5 wk of age, case 4 weighed 2801 gr and measured 64 cm. His mother noticed polyhydramnios developing and less fetal movement. He had weak swallowing, which made feeding difficult for him. When he was 11 months old, he could sit up on his own. By the time he was 2 yr old, he was walking and needed speech and language therapy. He could speak in full sentences by the time he finished primary school and was able to attend mainstream school with assistance. The following characteristics were indicative of dysmorphic features: forward nostrils, thick nasal alae, ptosis, prominent eyelashes, flat nasal bridge, short nose, broad nasal root and tip, and broad philtrum.
2.2.2. Genotype
A heterozygous de novo mutation in the glycosylphosphatidylinositol attachment to proteins inositol deacylase 1 (PGAP1) gene was detected by exome sequencing (NM_024989.4:c.1221-1G>T).
2.3. Case 5
2.3.1. Phenotype of the third family’s cases
Case 5 from the third family from Shiraz of Iran was born with a birth weight of 2987 gr and noted with stridor from birth. She was referred to the genetics clinic because of her short stature and moderate learning difficulties. Clinical examination revealed dysmorphic features, including downward-sloping palpebral fissures, low-set and backward-turned ears, a narrow and high palate, a small chin, and arachnodactyly. In addition, the 3rd and 4th toenails were hypoplastic. Generalized hypotonia has been observed in infancy. Skeletal abnormalities included mild pectus carinatum, thoracic hyperkyphosis, and lumbar hyperlordosis. At 7 months old, she underwent surgery for laryngitis. Although she was short in stature during her childhood, she reached a height of 154.5 cm in adulthood. At the age of 20 yr, she suffered from cerebral sinus thrombosis and was evaluated for homocystinuria, which was subsequently excluded. She is also reported to suffer from anxiety.
2.3.2. Genotype
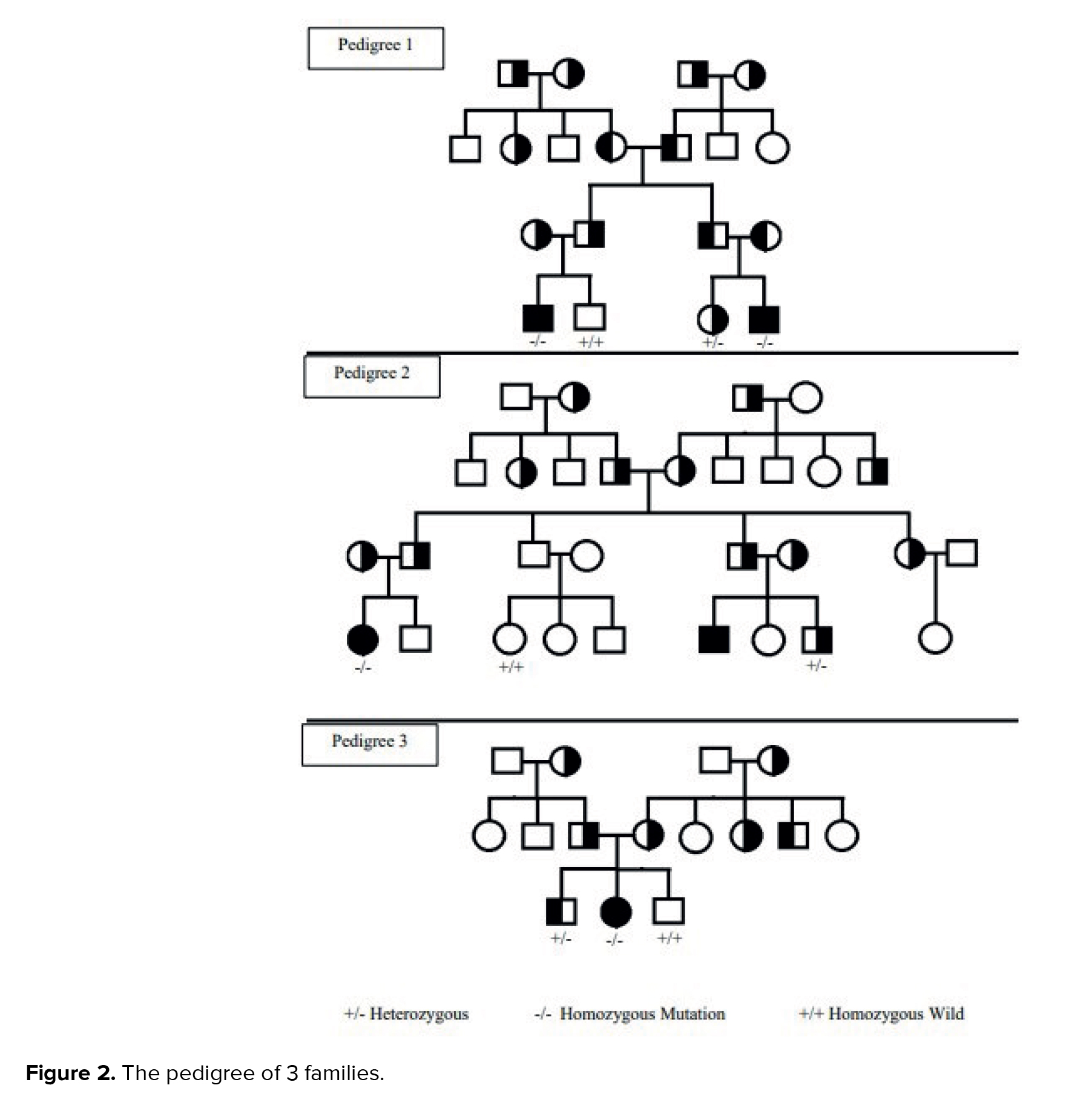
WES identified a homozygous de novo missense mutation in gene lysosomal trafficking regulator (LYST) (NM_000081.4:c.949G>A). The pedigree of 3 families (Figure 2) and the pathogenicity results of the discovered variants are explained in table II.

2.4. Ethical Considerations
This study was approved by the Ethics Committee of Islamic Azad University Varamin Branch, Tehran, Iran (Code: IR.IAU.VARAMIN.REC.1401.006). Informed consent, both written and oral, was obtained from all participants or their legal guardians prior to their inclusion in the study. Data confidentiality was strictly maintained, with measures in place to protect personal privacy and prevent unauthorized access to participant data.
3. Discussion
The application of WES allowed us to identify these novel mutations in genes not previously strongly associated with ID, highlighting the power of this technology in expanding our understanding of the genetic landscape of neurological disorders. PND plays a critical role in managing many genetic disorders, including mental retardation. Our study identified novel mutations in MDH1, PGAP1, and LYST genes in participants with ID, genes not previously strongly linked to ID. The identification of these mutations underscores the power of WES in uncovering the genetic basis of complex disorders. These genes are expressed in the brain and have roles in neuronal function (1).
Biallelic mutations in the LYST gene cause Chediak-Higashi syndrome (CHS), an autosomal recessive disorder characterized by abnormalities in lysosome-related organelles. While the precise function of lysosome-related organelles in human neuronal models remains an area of active investigation, studies have shed light on the role of LYST in neuronal function. For instance, Serra-Vinardell et al. demonstrated that LYST mutations can lead to lysosome depletion and the formation of hyper-elongated tubules in neurons. Their research, validated through the use of induced pluripotent stem cells derived from CHS individuals, revealed that LYST is essential for autolysosome tubule fission and fusion during autophagic lysosome reformation, a critical process for restoring free lysosomes following autophagy. LYST's localization to the lysosomal membrane suggests its involvement in facilitating the breakage of autolysosome tubules, underscoring its role in maintaining lysosomal homeostasis. These findings suggest a potential link between autophagic lysosome reformation dysregulation and neurodegeneration (10).
The LYST gene (HGNC: 1968), which spans 53 exons and produces a 13,503-bp mRNA transcript, encodes a protein crucial for sorting endosomal resident proteins into late multivesicular endosomes. To date, a significant number of pathogenic or likely pathogenic variants have been identified in the LYST gene, and investigations into genotype-phenotype relationships are ongoing for better understanding the clinical spectrum of CHS and related neurological manifestations (11). This highlights the importance of LYST in maintaining proper cellular function, particularly within the nervous system.
Brain aging involves energy metabolism dysfunction, including a decrease in tricarboxylic acid cycle variability. However, a large number of genes involved in this metabolic defect are still unknown. It is noteworthy that aging neurons have been found to exhibit downregulated levels of malate dehydrogenase, specifically MDH1 and MDH2. These 2 enzymes play a vital role in the malate-aspartate shuttle (MAS), which provides reducing equivalents for the electron transport chain and shuttles the transfer of NADH into the mitochondrial matrix (12). It has also been demonstrated that normal aging reduces the expression of MAS enzymes, though dietary restrictions can reverse this effect.
Additionally, children with severe neurological deficits are linked to loss-of-function mutations in the MDH genes. The significance of these enzymes in candidating brain age functional decline is highlighted by the NADH/NAD+ ratio, which is a crucial driver of the astrocyte-neuron-lactate shuttle (ANLS) along with pyruvate levels (13). Astrocytes play a vital role in neuronal energy metabolism through several key pathways. Primarily, they are responsible for aerobic glycolysis, converting pyruvate into lactate, which is then shuttled to neurons to fuel ATP production via oxidative phosphorylation (14). This process is part of the ANLS, a critical interaction that ensures neurons have sufficient energy for neurotransmission. In addition to the ANLS, astrocytes participate in the glutamate-glutamine cycle, where they uptake glutamate, the primary excitatory neurotransmitter, convert it to glutamine, and then return it to neurons for reconversion into glutamate. These interactions, along with the exchange of sodium and potassium ions, highlight the importance of astrocyte-neuron metabolic cooperation. Interestingly, brain metabolism becomes dysregulated during aging, with studies showing increased mitochondrial oxidative metabolism in astrocyte cultures from adult rats compared to young rats (15, 16).
MDH1 is a crucial enzyme in the MAS, which transports reducing equivalents across the mitochondrial membrane to maintain intracellular NAD(H) redox balance. Because NAD(H) cannot pass through the inner mitochondrial membrane, cytosolic and mitochondrial NAD+/NADH pools must remain independent to ensure proper redox balance for cell development and proliferation (17).
Studies on PGAP1 mutant mice have demonstrated DDs, male infertility, and forebrain development problems, including holoprosencephaly-like phenotypes. These mutants exhibit changes in the Wnt and Nodal signaling pathways, highlighting PGAP1's critical role in physiological functions and brain development (18). PGAP1 functions by remodeling GPI anchors on proteins after they are bound by GPI transamidase. As a GPI inositol deacylase, PGAP1 is located in the endoplasmic reticulum and removes the acyl chain from inositol (19).
Our findings, integrated with exome sequencing data and functional experiments, suggest a causative role for the identified splice acceptor mutation in PGAP1 in the severe, nonspecific autosomal recessive mental retardation observed in the studied family. This conclusion aligns with previous research linking ID to mutations in the GPI synthesis pathway and mouse models. Limitations of our study include the lack of in vivo or in vitro functional analyses to conclusively assess the pathogenicity of the identified variants. However, a comprehensive analysis, considering other genetic variations in these genes, suggests that the reported variants are likely causative. This highlights the need for further research to fully elucidate the roles of these genes in neuronal function and development (18).
4. Conclusion
This study underscores the transformative potential of PND in the context of ID and DD, demonstrating the power of WES to identify novel mutations in MDH1, PGAP1, and LYST across 3 Iranian families. Prenatal detection of these variants provides critical insights into the genetic underpinnings of ID/DD, enabling early management strategies, potentially reducing the disease burden, and informing reproductive decisions for families at risk. By validating the involvement of these genes in the pathophysiology of ID/DD, this research advocates for the broader integration of NGS technologies into prenatal care. Such integration would facilitate a shift from reactive clinical approaches to more proactive and preventive strategies, ultimately improving outcomes for affected individuals and their families.
Data Availability
Data supporting the findings of this study are available upon reasonable request from the corresponding author.
Author Contributions
V. Naseh and F. Tafvizi designed the study and conducted the research. N. Ghasemi, M.Y. Vahidi Mehrjardi, and H. Ashrafzadeh monitored, evaluated, and analyzed the results of the study. Further, V. Naseh, M.Y. Vahidi Mehrjardi, and H. Ashrafzadeh reviewed the article. All authors approved the final manuscript and take responsibility for the integrity of the data.
Acknowledgments
We are grateful to all family members who participated in this study. We thank the Yazd Reproductive Science Institute, Abortion Research Center, Shahid Sadoughi University of Medical Sciences, Yazd, Iran. Finally, we should mention that AI tools like Grammarly and Deep Seek were employed for tasks such as revision, and grammar checks.
Conflict of Interest
The other authors declare no conflict of interest.
Genomic DNA was extracted from peripheral blood (AddPrep Genomic DNA Extraction Kit; Korea), and its concentration and purity were measured using Thermo Scientific's NanoDrop 8000 spectrophotometer and Invitrogen's Qubit 2.0 fluorometer. Blood samples underwent genomic DNA isolation and cytogenetic procedures following standard protocols. Peripheral blood lymphocytes were cultured, and chromosome staining was performed using G-banding as per Seabright’s methodology (1971).
Both Splice Site Prediction and Human Splicing Finder tools were employed to assess the impact of splice variants on transcripts. In line with American College of Medical Genetics and Genomics guidelines, variants were categorized as "pathogenic", "probably pathogenic", "uncertain significance", "probably benign", or "benign". Sanger sequencing was performed on index cases and other family members to confirm the identified variants.
All cases, including 5 cases from 3 families, underwent WES. Variants were annotated using the variant effect predictor and filtered based on the following criteria: 1) nonsynonymous single nucleotide variants or indels located in exon or splice regions; 2) absent in control populations or with an allele frequency < 0.01 in the exome sequencing project, 1000G, ExAC or genome aggregation database (gnomAD); 3) likely to be harmful from at least 4 of the following 6 software tools: SIFT, PolyPhen-2, MutationTaster (7), CADD (8), M-CAP (9), and Condel and PROVEAN. Splice site prediction along with human splicing finder were used to estimate the impact of splice variants on transcripts. In accordance with American College of Medical Genetics and Genomics guidelines and pertinent annotation files, variants were categorized as "pathogenic", "probably pathogenic", "uncertain significance", "probably benign", or "benign". To verify the validity and distinguish between potential variants, Sanger sequencing was carried out on the index participants as well as additional family members (Figure 1).
Using the following standards, the identified variants' clinical impact was assessed: 1) a frequency of alleles < 0.03 in the 1000G or exome variant server, 2) splicing, stop-gain, and frameshift variants were all considered to be highly pathogenic, 3) for missense mutations: assessments comprised Grantham scores, SIFT, PolyPhen-2, and amino acid conservation for pathogenicity predictions, 4) various patterns of inheritance, such as dominant inheritance, recessive inheritance, or maternal inheritance of X-linked variants, 5) variant absence in other dataset (in-house database), 6) evaluate the possible influence of synonymous or intronic variants the human splicing finder web tool was utilized. Furthermore, the pathogenicity of de novo variant was further demonstrated by the use of conservation restriction statistics for every gene from the ExAC dataset (http://exac.broadinstitute.org) in conjunction with the CADD score.
Sanger sequencing verified relevant genetic variants. After the cases, his parents, and a few other family members' DNA were amplified by polymerase chain reaction using particular primers, bidirectional sequencing was carried out on an Applied Biosystem (ABI PRISM 3500) automated sequencer (life technologies) using the BigDye Terminator kit v1.1. Exon primer was used to create all the primers needed for amplification and sequencing. On request, comprehensive primer sequences and polymerase chain reactionconditions can be obtained (Table I).
2.1. Cases 1 and 2
2.1.1. Phenotype of the first family’s cases
The first family from Zahedan, Iran, comprised cases 1 and 2. Case 1 weighed 3050 gr at birth. At the age of 6 months, this participant was referred to the clinical genetics department to measure disproportionate growth parameters and growth delay.
Cranial ultrasound results were normal. At the age of 2, he weighed 14 kg and was 79 cm tall. He was classified as moderately delayed in all aspects of development, could sit independently at 11 months, and started walking between 2 and 3 yr of age. He started talking at about 6 yr old and could say simple words and was also able to follow one-step commands. This boy was sensitive to loud noises and crowded places. In the examination, at the age of 6 yr and 10 months, his height was 100 cm, and his weight reached 21 kg, which indicated relative macrocephaly. He had intermittent strabismus but was visually quite satisfactory. Unlike his fingernails, which were normal, all his toenails, especially the 4th and 5th toenails, were hypoplastic. The results of the MRI scan of the skull at the age of 9 months showed a corpus callosum with mild deformity, which delayed myelination.
Case no. 2 was referred to the clinical genetics center due to his growth and development.
He weighed 2.65 kg at birth. He had laryngomalacia and had problems such as feeding difficulty, vomiting, constipation, and sleep disturbance. The results of the developmental evaluation showed that he has a moderate DD (he started to walk at 22 months and started his first words at 20 months). This boy was short (91.8 cm) with a normal head circumference and had facial deformities, including a thin upper lip and micrognathia.
2.1.2. Genotype
In the malate dehydrogenase 1 (MDH1) gene, a homozygous de novo stop-gain variant was found, specifically (NM_005917.4:c.4C>T). The truncation of the MDH1 protein occurs due to the introduction of an early stop codon caused by the mentioned base change.
2.2. Cases 3 and 4
2.2.1. Phenotype of the second family’s cases
Cases number 3 and 4 were from Yazd, Iran. Fetal scans revealed renal pelvic enlargement in case 3. She weighed 3100 gr at birth. She had severe feeding issues and DDS in her early years; at 14 wk, she smiled, at 13 months, she could sit by herself, and at 23 months, she could walk by herself. She was observed to be utilizing single words at the age of 2, but her primary mode of communication was nonverbal. She weighed 11.2 kg and was 87 cm tall. She experienced frequent infections and constipation as a baby. Myotonic dystrophy was ruled out, and a clinical examination revealed generalized hypotension, joint hypermobility, and slightly elevated creatine kinase levels. On assessment, she also had plagiocephaly, prominent ears, a thin upper lip of vermilion, and downturned mouth corners.
At 5 wk of age, case 4 weighed 2801 gr and measured 64 cm. His mother noticed polyhydramnios developing and less fetal movement. He had weak swallowing, which made feeding difficult for him. When he was 11 months old, he could sit up on his own. By the time he was 2 yr old, he was walking and needed speech and language therapy. He could speak in full sentences by the time he finished primary school and was able to attend mainstream school with assistance. The following characteristics were indicative of dysmorphic features: forward nostrils, thick nasal alae, ptosis, prominent eyelashes, flat nasal bridge, short nose, broad nasal root and tip, and broad philtrum.
2.2.2. Genotype
A heterozygous de novo mutation in the glycosylphosphatidylinositol attachment to proteins inositol deacylase 1 (PGAP1) gene was detected by exome sequencing (NM_024989.4:c.1221-1G>T).
2.3. Case 5
2.3.1. Phenotype of the third family’s cases
Case 5 from the third family from Shiraz of Iran was born with a birth weight of 2987 gr and noted with stridor from birth. She was referred to the genetics clinic because of her short stature and moderate learning difficulties. Clinical examination revealed dysmorphic features, including downward-sloping palpebral fissures, low-set and backward-turned ears, a narrow and high palate, a small chin, and arachnodactyly. In addition, the 3rd and 4th toenails were hypoplastic. Generalized hypotonia has been observed in infancy. Skeletal abnormalities included mild pectus carinatum, thoracic hyperkyphosis, and lumbar hyperlordosis. At 7 months old, she underwent surgery for laryngitis. Although she was short in stature during her childhood, she reached a height of 154.5 cm in adulthood. At the age of 20 yr, she suffered from cerebral sinus thrombosis and was evaluated for homocystinuria, which was subsequently excluded. She is also reported to suffer from anxiety.
2.3.2. Genotype
WES identified a homozygous de novo missense mutation in gene lysosomal trafficking regulator (LYST) (NM_000081.4:c.949G>A). The pedigree of 3 families (Figure 2) and the pathogenicity results of the discovered variants are explained in table II.
2.4. Ethical Considerations
This study was approved by the Ethics Committee of Islamic Azad University Varamin Branch, Tehran, Iran (Code: IR.IAU.VARAMIN.REC.1401.006). Informed consent, both written and oral, was obtained from all participants or their legal guardians prior to their inclusion in the study. Data confidentiality was strictly maintained, with measures in place to protect personal privacy and prevent unauthorized access to participant data.
3. Discussion
The application of WES allowed us to identify these novel mutations in genes not previously strongly associated with ID, highlighting the power of this technology in expanding our understanding of the genetic landscape of neurological disorders. PND plays a critical role in managing many genetic disorders, including mental retardation. Our study identified novel mutations in MDH1, PGAP1, and LYST genes in participants with ID, genes not previously strongly linked to ID. The identification of these mutations underscores the power of WES in uncovering the genetic basis of complex disorders. These genes are expressed in the brain and have roles in neuronal function (1).
Biallelic mutations in the LYST gene cause Chediak-Higashi syndrome (CHS), an autosomal recessive disorder characterized by abnormalities in lysosome-related organelles. While the precise function of lysosome-related organelles in human neuronal models remains an area of active investigation, studies have shed light on the role of LYST in neuronal function. For instance, Serra-Vinardell et al. demonstrated that LYST mutations can lead to lysosome depletion and the formation of hyper-elongated tubules in neurons. Their research, validated through the use of induced pluripotent stem cells derived from CHS individuals, revealed that LYST is essential for autolysosome tubule fission and fusion during autophagic lysosome reformation, a critical process for restoring free lysosomes following autophagy. LYST's localization to the lysosomal membrane suggests its involvement in facilitating the breakage of autolysosome tubules, underscoring its role in maintaining lysosomal homeostasis. These findings suggest a potential link between autophagic lysosome reformation dysregulation and neurodegeneration (10).
The LYST gene (HGNC: 1968), which spans 53 exons and produces a 13,503-bp mRNA transcript, encodes a protein crucial for sorting endosomal resident proteins into late multivesicular endosomes. To date, a significant number of pathogenic or likely pathogenic variants have been identified in the LYST gene, and investigations into genotype-phenotype relationships are ongoing for better understanding the clinical spectrum of CHS and related neurological manifestations (11). This highlights the importance of LYST in maintaining proper cellular function, particularly within the nervous system.
Brain aging involves energy metabolism dysfunction, including a decrease in tricarboxylic acid cycle variability. However, a large number of genes involved in this metabolic defect are still unknown. It is noteworthy that aging neurons have been found to exhibit downregulated levels of malate dehydrogenase, specifically MDH1 and MDH2. These 2 enzymes play a vital role in the malate-aspartate shuttle (MAS), which provides reducing equivalents for the electron transport chain and shuttles the transfer of NADH into the mitochondrial matrix (12). It has also been demonstrated that normal aging reduces the expression of MAS enzymes, though dietary restrictions can reverse this effect.
Additionally, children with severe neurological deficits are linked to loss-of-function mutations in the MDH genes. The significance of these enzymes in candidating brain age functional decline is highlighted by the NADH/NAD+ ratio, which is a crucial driver of the astrocyte-neuron-lactate shuttle (ANLS) along with pyruvate levels (13). Astrocytes play a vital role in neuronal energy metabolism through several key pathways. Primarily, they are responsible for aerobic glycolysis, converting pyruvate into lactate, which is then shuttled to neurons to fuel ATP production via oxidative phosphorylation (14). This process is part of the ANLS, a critical interaction that ensures neurons have sufficient energy for neurotransmission. In addition to the ANLS, astrocytes participate in the glutamate-glutamine cycle, where they uptake glutamate, the primary excitatory neurotransmitter, convert it to glutamine, and then return it to neurons for reconversion into glutamate. These interactions, along with the exchange of sodium and potassium ions, highlight the importance of astrocyte-neuron metabolic cooperation. Interestingly, brain metabolism becomes dysregulated during aging, with studies showing increased mitochondrial oxidative metabolism in astrocyte cultures from adult rats compared to young rats (15, 16).
MDH1 is a crucial enzyme in the MAS, which transports reducing equivalents across the mitochondrial membrane to maintain intracellular NAD(H) redox balance. Because NAD(H) cannot pass through the inner mitochondrial membrane, cytosolic and mitochondrial NAD+/NADH pools must remain independent to ensure proper redox balance for cell development and proliferation (17).
Studies on PGAP1 mutant mice have demonstrated DDs, male infertility, and forebrain development problems, including holoprosencephaly-like phenotypes. These mutants exhibit changes in the Wnt and Nodal signaling pathways, highlighting PGAP1's critical role in physiological functions and brain development (18). PGAP1 functions by remodeling GPI anchors on proteins after they are bound by GPI transamidase. As a GPI inositol deacylase, PGAP1 is located in the endoplasmic reticulum and removes the acyl chain from inositol (19).
Our findings, integrated with exome sequencing data and functional experiments, suggest a causative role for the identified splice acceptor mutation in PGAP1 in the severe, nonspecific autosomal recessive mental retardation observed in the studied family. This conclusion aligns with previous research linking ID to mutations in the GPI synthesis pathway and mouse models. Limitations of our study include the lack of in vivo or in vitro functional analyses to conclusively assess the pathogenicity of the identified variants. However, a comprehensive analysis, considering other genetic variations in these genes, suggests that the reported variants are likely causative. This highlights the need for further research to fully elucidate the roles of these genes in neuronal function and development (18).
4. Conclusion
This study underscores the transformative potential of PND in the context of ID and DD, demonstrating the power of WES to identify novel mutations in MDH1, PGAP1, and LYST across 3 Iranian families. Prenatal detection of these variants provides critical insights into the genetic underpinnings of ID/DD, enabling early management strategies, potentially reducing the disease burden, and informing reproductive decisions for families at risk. By validating the involvement of these genes in the pathophysiology of ID/DD, this research advocates for the broader integration of NGS technologies into prenatal care. Such integration would facilitate a shift from reactive clinical approaches to more proactive and preventive strategies, ultimately improving outcomes for affected individuals and their families.
Data Availability
Data supporting the findings of this study are available upon reasonable request from the corresponding author.
Author Contributions
V. Naseh and F. Tafvizi designed the study and conducted the research. N. Ghasemi, M.Y. Vahidi Mehrjardi, and H. Ashrafzadeh monitored, evaluated, and analyzed the results of the study. Further, V. Naseh, M.Y. Vahidi Mehrjardi, and H. Ashrafzadeh reviewed the article. All authors approved the final manuscript and take responsibility for the integrity of the data.
Acknowledgments
We are grateful to all family members who participated in this study. We thank the Yazd Reproductive Science Institute, Abortion Research Center, Shahid Sadoughi University of Medical Sciences, Yazd, Iran. Finally, we should mention that AI tools like Grammarly and Deep Seek were employed for tasks such as revision, and grammar checks.
Conflict of Interest
The other authors declare no conflict of interest.
Type of Study: Case Report |
Subject:
Reproductive Genetics
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
