
International Journal of
Reproductive Biomedicine
Sun, Jul 26, 2026
[Archive]
Volume 23, Issue 12 (December 2025)
IJRM 2025, 23(12): 1043-1050 |
Back to browse issues page



![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Haj Mohammad Hassani B, Ghasemi N, Malekzadeh K. A novel homozygous growth differentiation factor 9 variant associated with premature ovarian insufficiency: A case report. IJRM 2025; 23 (12) :1043-1050
URL: http://ijrm.ir/article-1-3709-en.html
URL: http://ijrm.ir/article-1-3709-en.html



1- Department of Medical Genetics, Faculty of Medicine, Hormozgan University of Medical Sciences, Bandar Abbas, Iran.
2- Department of Medical Genetics, Faculty of Medicine, Hormozgan University of Medical Sciences, Bandar Abbas, Iran. & Molecular Medicine Research Center, Hormozgan Health Institute, Hormozgan University of Medical Sciences, Bandar Abbas, Iran. ,keyanoosh@gmail.com; kianoosh.malekzadeh@hums.ac.ir
2- Department of Medical Genetics, Faculty of Medicine, Hormozgan University of Medical Sciences, Bandar Abbas, Iran. & Molecular Medicine Research Center, Hormozgan Health Institute, Hormozgan University of Medical Sciences, Bandar Abbas, Iran. ,
Full-Text [PDF 1718 kb]
(407 Downloads)
| Abstract (HTML) (532 Views)
1. Introduction
Premature ovarian insufficiency (POI) is a condition characterized by altered ovarian function, usually diagnosed in women during the second or third decade of life (1). Recent research has provided valuable information regarding its etiology, clinical features, management options, and impact on quality of life. The prevalence of POI is approximately 1-3% among women under the age of 40, but the condition's etiology remains complex and mostly unknown (2).
The major clinical manifestations of POI are amenorrhea for at least 4 months, increased levels of gonadotropins, most particularly follicle-stimulating hormone (FSH), and hypoestrogenism.
Individuals with POI often experience symptoms such as hot flashes and night sweats (1, 2). The diagnosis of POI usually requires clinical evaluation and laboratory tests to provide an initial differential diagnosis from hypogonadotropic hypogonadism, polycystic ovary syndrome, and related disorders. Transvaginal ultrasonography may also reveal small and inactive ovaries or a depleted follicle reserve (3, 4).
Autoimmune disorders and genetic abnormalities, such as turner and fragile X syndromes, are the well-known causes of POI (5). Additionally, variants in other genes involved in follicle development, such as growth differentiation factor 9 (GDF9), have been associated with POI. GDF9 encodes a member of the transforming growth factor-beta superfamily secreted by oocytes, which plays a key role in folliculogenesis. Initial studies indicated autosomal dominant inheritance for GDF9-related POI, whereas recent evidence suggested an autosomal recessive pattern (6). Our study investigated the genetic basis of POI in an Iranian family with 2 affected sisters using whole-exome sequencing (WES). Our findings highlight the potential autosomal recessive inheritance of POI caused by GDF9 variants, and further expand the mutational spectrum of this gene associated with the disease.
2. Case Presentation
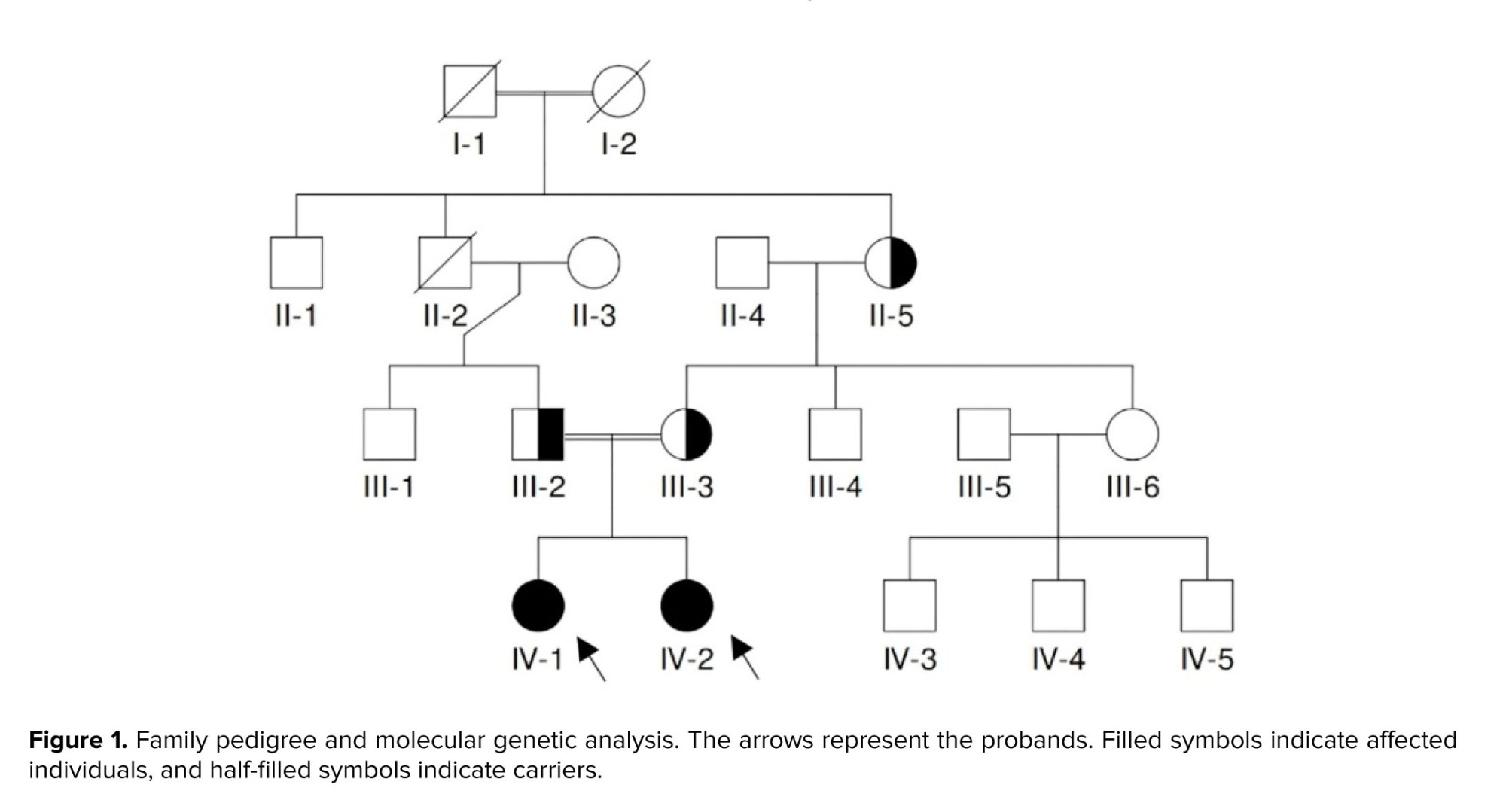
We investigated 2 POI-affected sisters (IV-1 and IV-2) (Figure 1), who were referred to Shahid Mohammadi hospital, Bandar Abbas, Iran.
They experienced menarche at ages 14 and 13 and subsequently developed secondary amenorrhea at ages 17 and 15, respectively. The physical examination revealed normal external genitalia and secondary sexual characteristics. Hormonal evaluations demonstrated elevated gonadotropins in participants IV-1 (FSH = 61.3 IU/L, luteinizing hormone [LH] = 37.4 IU/L) and IV-2 (FSH = 77.1 IU/L, LH = 43.8 IU/L), with low estradiol levels and absent anti-Müllerian hormone levels, confirming a hypergonadotropic hypogonadism profile. The ultrasonography revealed small ovaries in both cases. In case IV-1, the left ovary measured 11 × 12 × 4 mm and the right ovary 16 × 14 × 6 mm. In case IV-2, the left ovary measured 16 × 11 × 4 mm and the right ovary 18 × 15 × 5 mm. Antral follicles were absent in both probands, while uterine volumes remained within normal limits. Additional laboratory investigations excluded other common causes of POI. The laboratory evaluations of thyroid function, together with adrenal hormone profiles, demonstrated normal results, and cytogenetic analysis revealed normal female karyotypes (46, XX) in the participants. The molecular examination for the fragile X mental retardation-1 premutation also has a negative outcome. Hormone therapy was initiated for them, consisting of 10 mg medroxyprogesterone acetate for 14 days per month and 1.25 mg conjugated equine estrogen for 25 days per month. This regimen, together with vitamin D and calcium supplementation, effectively managed their symptoms.
WES was conducted for both affected sisters. The SureSelectQXT kit (Agilent) was used for library preparation, and paired-end sequencing with 100x coverage was performed on the NovaSeq 6000 (Illumina, San Diego, CA, USA). The FastQC tool (v.0.11.9) was used to evaluate the quality of the Fastq data, and genomic alignment against the hg19 assembly was performed using BWA-MEM (v.0.7.17). The variants were called GATK HaplotypeCaller (v.4.4) and then annotated with ANNOVAR. Variants were filtered based on minor allele frequency (MAF < 1%) and American College of Medical Genetics and Genomics guidelines, with only high-confidence variants selected for final interpretation. The ClinVar, Franklin, Varsome, and InterVare databases were used to examine variant classification, and the OMIM, DisGeNET, and GeneCards databases were used to investigate the gene-disease associations.
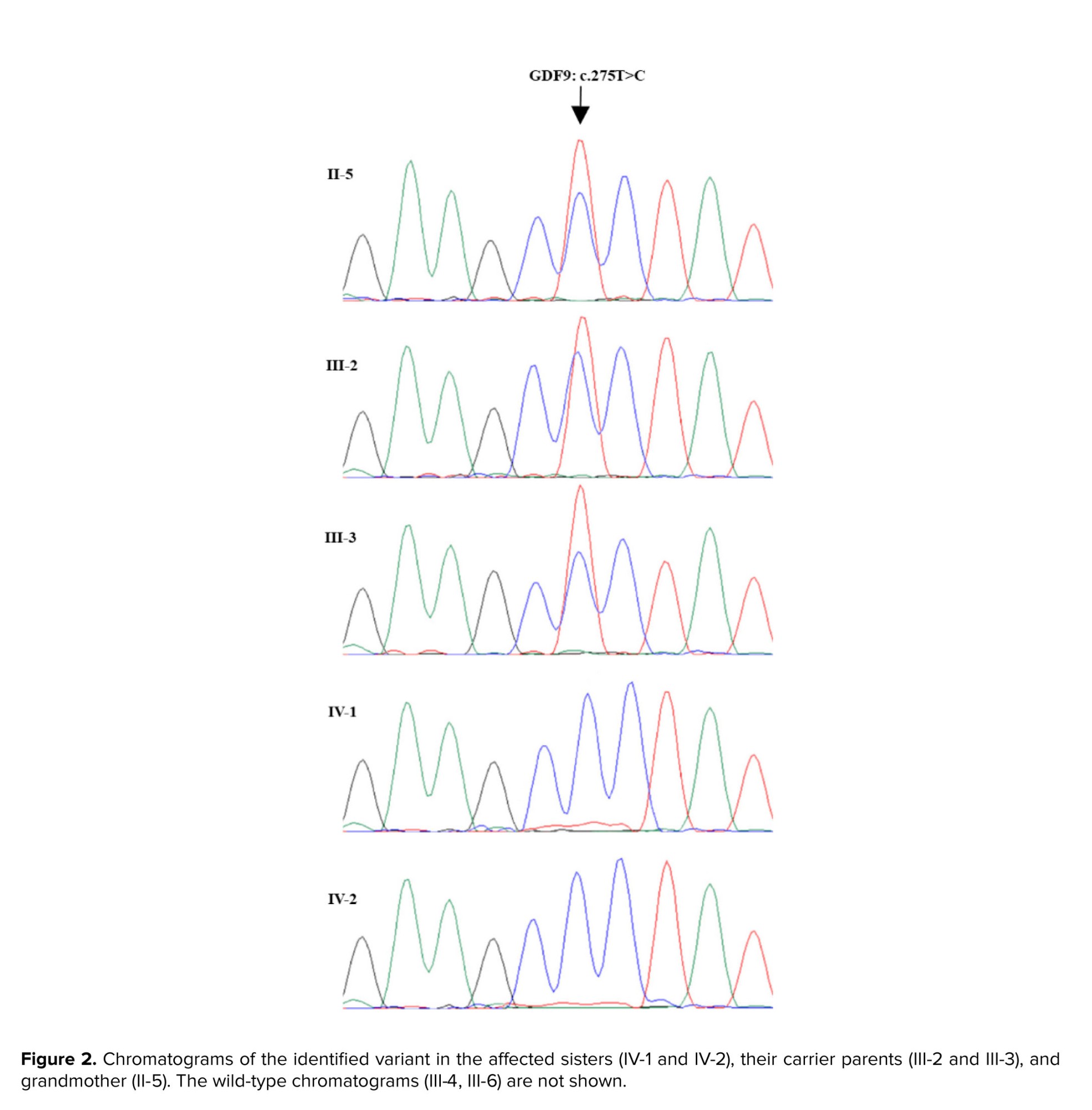
WES identified a novel homozygous GDF9 gene variant (NM_005260.6: c.275T>C; Leu92Pro) in the POI-affected sisters. PCR amplification (95°C for 5 min, followed by 28 cycles at 95°C for 30 sec, 58°C for 45 sec, 72°C for 30 sec, and 72°C for 5 min), Sanger sequencing, and subsequent segregation analysis were conducted using specifically designed forward 5′-TTTTCCTATTAGCCTTGGTTCTC-3′ and reverse 5′-CACATACCTGTTACCTGGTCTCC-3′ primers (Figures 1 and 2).
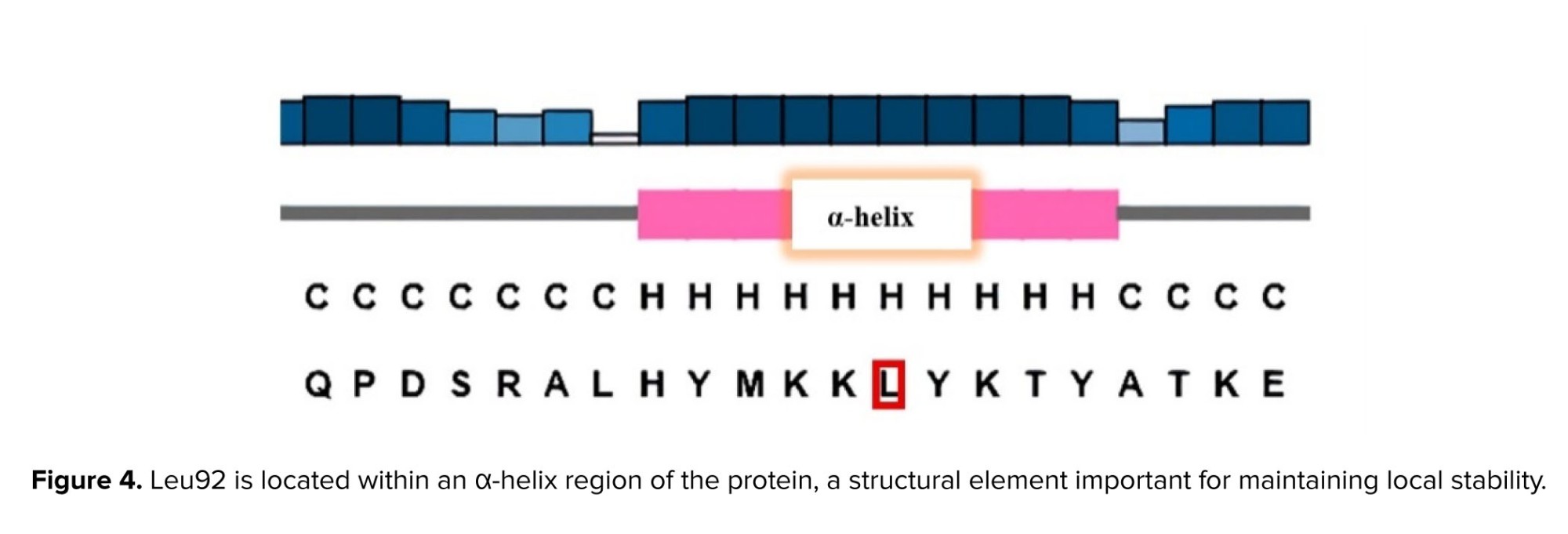
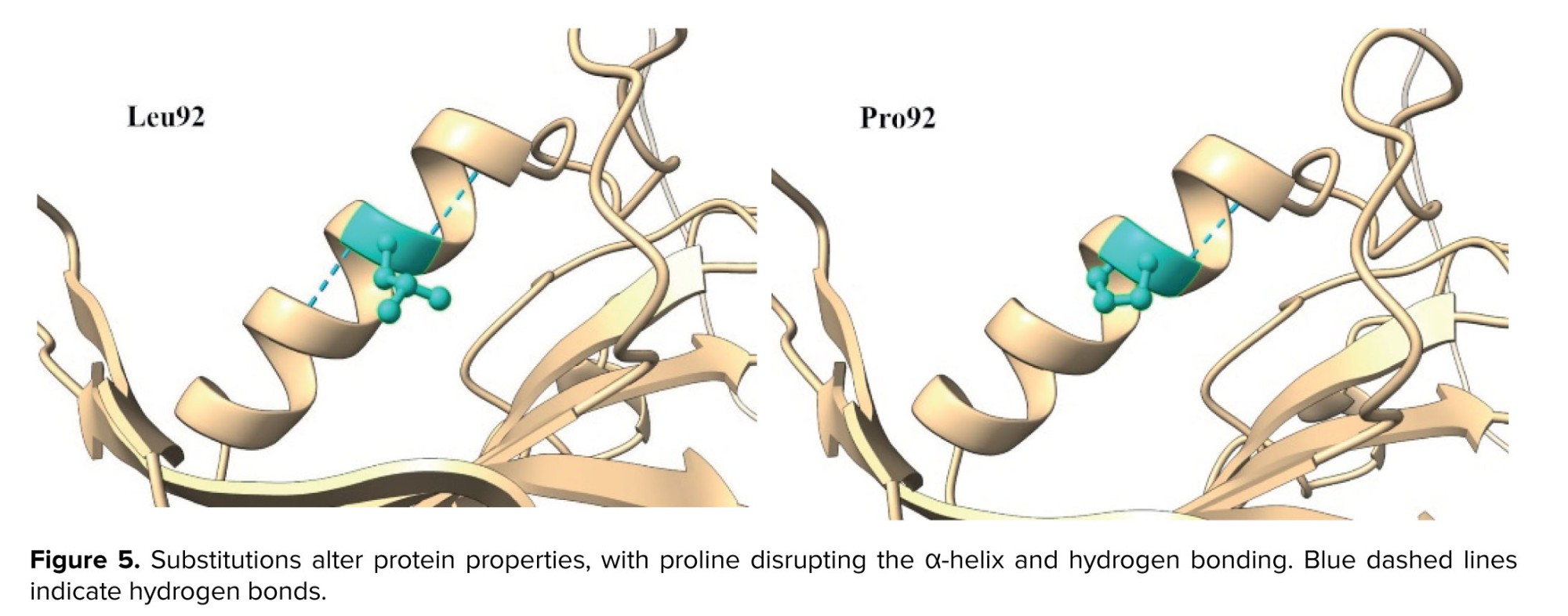
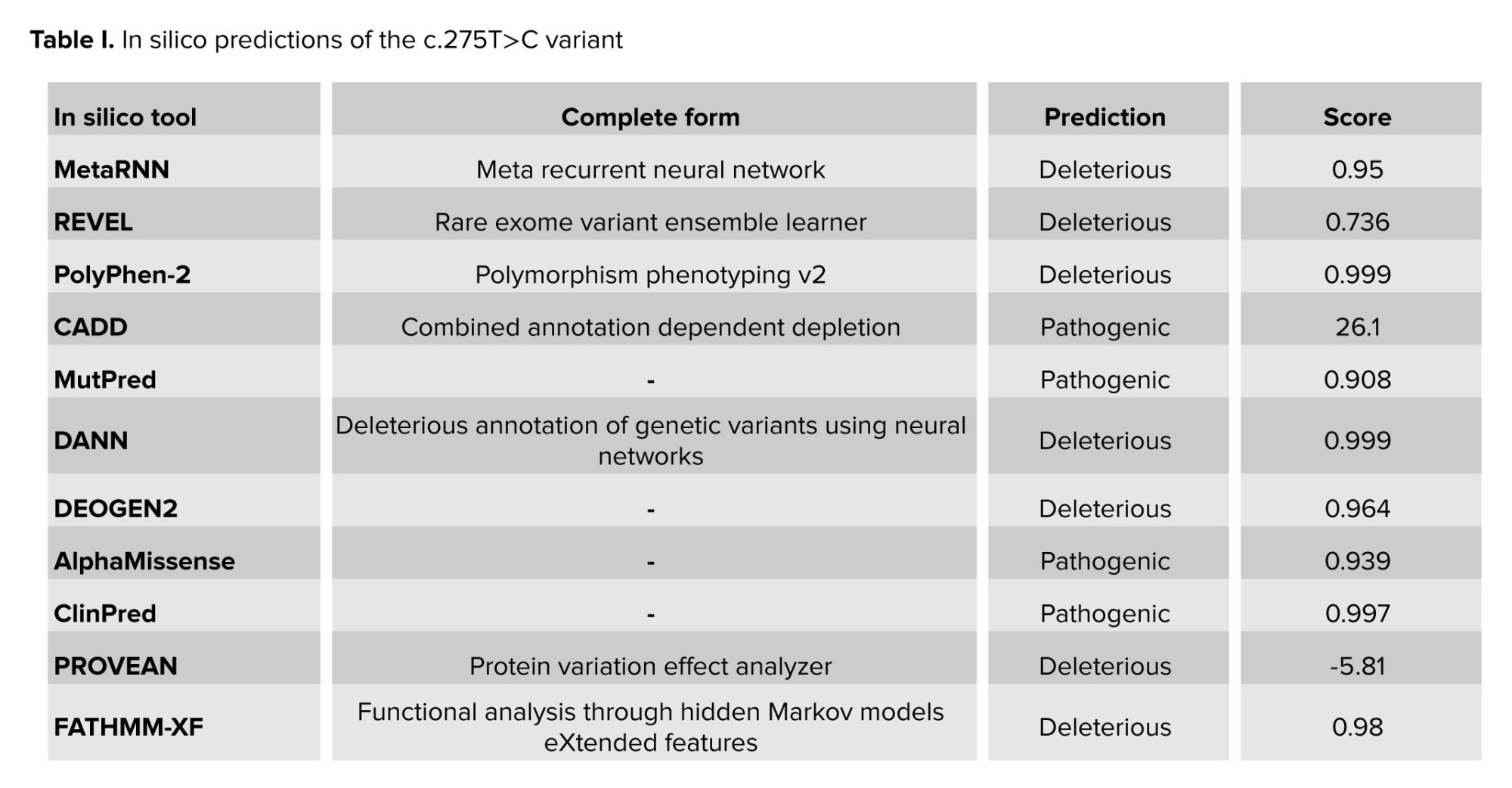
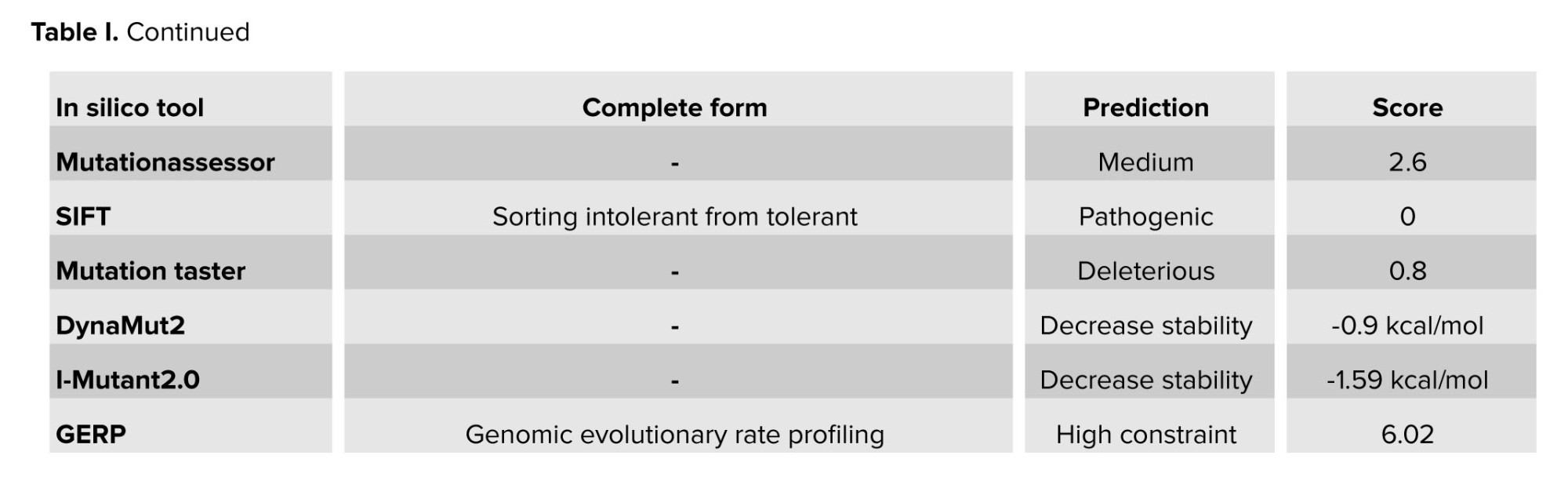
The variant was observed to be absent from the Iranome database (https://iranome.com) and Genome Aggregation Database v2.1.1 (gnomAD v2.1.1) (https://gnomad.broadinstitute.org). Based on American College of Medical Genetics and Genomics criteria, the c.275T>C variant was categorized as a variant of uncertain significance, supported by PM1, PM2, and PP3 evidence. Analyses using various in-silico tools, such as I-mutant2.0 and MutationTaster, predicted the effect of the variant (Table I). Conservation assessment was performed using ConSurf and Clustal Omega (Figure 3). Structural prediction of the region containing Leu92 was generated using the PSIPRED database (https://bioinf.cs.ucl.ac.uk) (Figure 4), and 3-dimensional molecular modeling was conducted with Chimera software (Figure 5).

2.1. Ethical Considerations
Written informed consent was obtained from both cases in this study.
3. Discussion
POI manifests through ovarian dysfunction and menopause-like symptoms, while genetic defects and autoimmune disorders serve as the main causes for its development. However, the underlying reason remains unidentified in more than half of POI cases (7).
In this study, we describe a family where 2 sisters were affected by early secondary amenorrhea before age 20. WES analysis identified a homozygous GDF9 variant (c.275T>C; Leu92Pro) in the probands, whereas their parents and grandmother were heterozygous carriers. Neither the carrier mother nor the grandmother exhibited clinical features of POI or premature menopause. Their mother and grandmother experienced menopause at ages 49 and 55, respectively. The variant was absent in population databases, and in silico analyses predicted its deleterious effects. According to the PSIPRED database, leucine at position 92 was located within an α-helix. Therefore, the Leu92Pro substitution was expected to disrupt local protein structure and α-helix formation.
GDF9 functions as an oocyte-produced growth factor that plays a crucial role in the initial stages of follicle development and in maintaining ovarian health. Different genetic variations in the GDF9 gene show association with both poor follicle development and diminished ovarian reserve, alongside cases of familial POI. Functional studies indicate that several GDF9 missense variants impair protein maturation and reduce the biological activity of conditioned media from HEK293F cells compared with the wild-type protein (8).
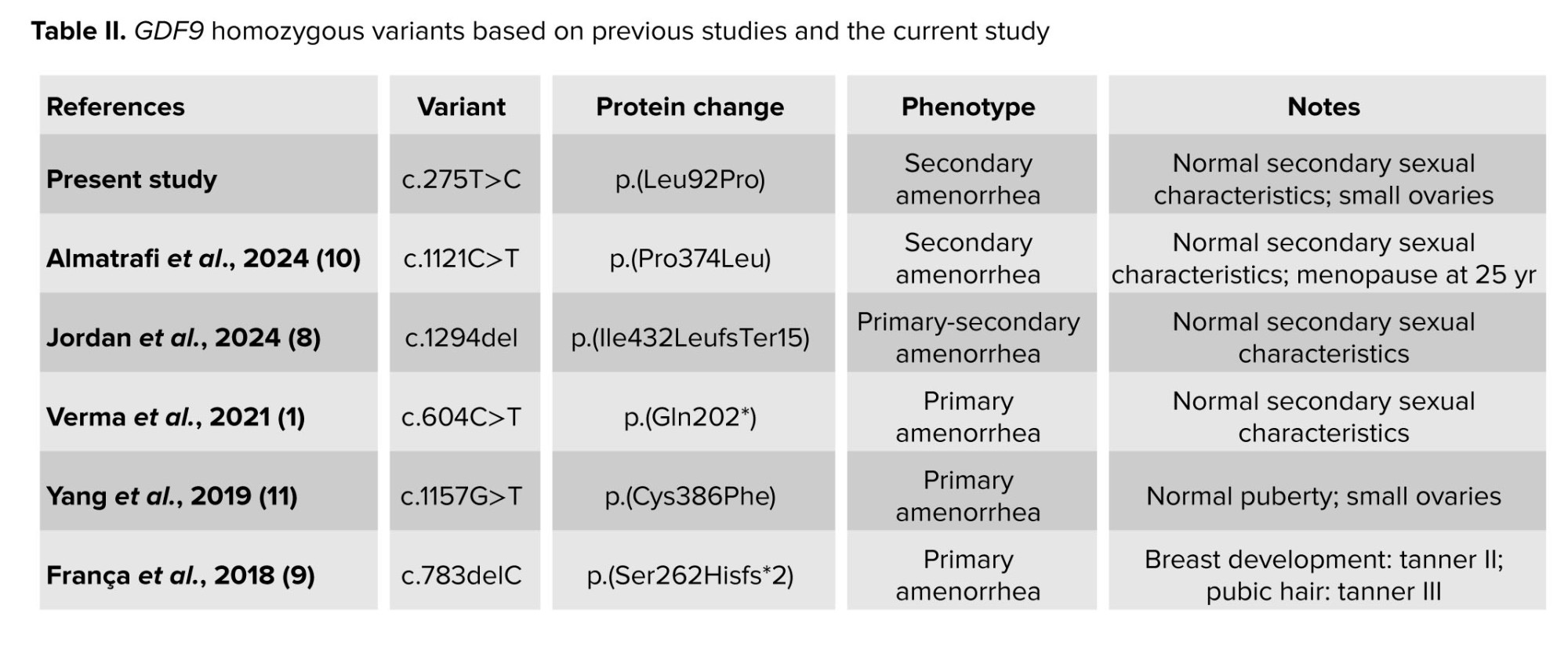
Initial cohort studies suggested that heterozygous GDF9 variants may cause POI in an autosomal dominant manner. Studies in French, Chinese, and Indian cohorts identified several heterozygous missense variants, including p.Ser186Tyr, p.Pro103Ser, p.Thr238Ala, p.Lys67Glu, and p.Val216Met in individuals with POI. Most variants were rare, and their pathogenicity remained uncertain. However, functional assays demonstrated the effect of missense variants, such as p.Lys67Glu and p.Pro103Ser, on protein function. In contrast, recent studies indicate that heterozygous variants have a milder effect. These studies suggested autosomal recessive inheritance of the disease due to GDF9 variants. They also claimed that heterozygous variants of this gene are associated with normal ovarian function or secondary amenorrhea at older ages (1, 8-11) (Table II).
The normal phenotypes of the mother and grandmother of our study proband further support the recessive mode of inheritance. It was observed that the clinical severity of POI is influenced by both the number of affected GDF9 alleles and the specific nature of the mutations. While a single heterozygous missense variant may play a contributory role in the onset of premature menopause, it may not necessarily lead to early menopause in all carriers. In contrast, compound heterozygous or homozygous variants that were experimentally confirmed or predicted to result in substantial impairment of protein function were more likely to cause POI or early amenorrhea.
4. Conclusion
In conclusion, we report a homozygous variant in GDF9 co-segregating with POI in affected sisters, providing support for a Mendelian gene-disease association. This finding also expands the known mutational spectrum of GDF9 and underscores its importance for genetic counseling and molecular diagnosis in families affected by POI.
Author Contributions
B. Haj Mohammad Hassani: Conceptualization, bioinformatics analysis, literature review, and article writing. N. Ghasemi: Literature review and article writing. K. Malekzadeh: Data collection, supervision, editing, and review.
Acknowledgments
The researchers sincerely appreciate both the cases and the health service staff of Shahid Mohammadi hospital, Bandar Abbas, Iran who contributed to this research. The authors declare that no funds, grants, or other support were received during the preparation of this manuscript. The authors of this article did not use artificial intelligence (AI) in any way.
Conflict of Interest
The authors declare that there is no conflict of interest.
Full-Text: (74 Views)
1. Introduction
Premature ovarian insufficiency (POI) is a condition characterized by altered ovarian function, usually diagnosed in women during the second or third decade of life (1). Recent research has provided valuable information regarding its etiology, clinical features, management options, and impact on quality of life. The prevalence of POI is approximately 1-3% among women under the age of 40, but the condition's etiology remains complex and mostly unknown (2).
The major clinical manifestations of POI are amenorrhea for at least 4 months, increased levels of gonadotropins, most particularly follicle-stimulating hormone (FSH), and hypoestrogenism.
Individuals with POI often experience symptoms such as hot flashes and night sweats (1, 2). The diagnosis of POI usually requires clinical evaluation and laboratory tests to provide an initial differential diagnosis from hypogonadotropic hypogonadism, polycystic ovary syndrome, and related disorders. Transvaginal ultrasonography may also reveal small and inactive ovaries or a depleted follicle reserve (3, 4).
Autoimmune disorders and genetic abnormalities, such as turner and fragile X syndromes, are the well-known causes of POI (5). Additionally, variants in other genes involved in follicle development, such as growth differentiation factor 9 (GDF9), have been associated with POI. GDF9 encodes a member of the transforming growth factor-beta superfamily secreted by oocytes, which plays a key role in folliculogenesis. Initial studies indicated autosomal dominant inheritance for GDF9-related POI, whereas recent evidence suggested an autosomal recessive pattern (6). Our study investigated the genetic basis of POI in an Iranian family with 2 affected sisters using whole-exome sequencing (WES). Our findings highlight the potential autosomal recessive inheritance of POI caused by GDF9 variants, and further expand the mutational spectrum of this gene associated with the disease.
2. Case Presentation
We investigated 2 POI-affected sisters (IV-1 and IV-2) (Figure 1), who were referred to Shahid Mohammadi hospital, Bandar Abbas, Iran.
They experienced menarche at ages 14 and 13 and subsequently developed secondary amenorrhea at ages 17 and 15, respectively. The physical examination revealed normal external genitalia and secondary sexual characteristics. Hormonal evaluations demonstrated elevated gonadotropins in participants IV-1 (FSH = 61.3 IU/L, luteinizing hormone [LH] = 37.4 IU/L) and IV-2 (FSH = 77.1 IU/L, LH = 43.8 IU/L), with low estradiol levels and absent anti-Müllerian hormone levels, confirming a hypergonadotropic hypogonadism profile. The ultrasonography revealed small ovaries in both cases. In case IV-1, the left ovary measured 11 × 12 × 4 mm and the right ovary 16 × 14 × 6 mm. In case IV-2, the left ovary measured 16 × 11 × 4 mm and the right ovary 18 × 15 × 5 mm. Antral follicles were absent in both probands, while uterine volumes remained within normal limits. Additional laboratory investigations excluded other common causes of POI. The laboratory evaluations of thyroid function, together with adrenal hormone profiles, demonstrated normal results, and cytogenetic analysis revealed normal female karyotypes (46, XX) in the participants. The molecular examination for the fragile X mental retardation-1 premutation also has a negative outcome. Hormone therapy was initiated for them, consisting of 10 mg medroxyprogesterone acetate for 14 days per month and 1.25 mg conjugated equine estrogen for 25 days per month. This regimen, together with vitamin D and calcium supplementation, effectively managed their symptoms.
WES was conducted for both affected sisters. The SureSelectQXT kit (Agilent) was used for library preparation, and paired-end sequencing with 100x coverage was performed on the NovaSeq 6000 (Illumina, San Diego, CA, USA). The FastQC tool (v.0.11.9) was used to evaluate the quality of the Fastq data, and genomic alignment against the hg19 assembly was performed using BWA-MEM (v.0.7.17). The variants were called GATK HaplotypeCaller (v.4.4) and then annotated with ANNOVAR. Variants were filtered based on minor allele frequency (MAF < 1%) and American College of Medical Genetics and Genomics guidelines, with only high-confidence variants selected for final interpretation. The ClinVar, Franklin, Varsome, and InterVare databases were used to examine variant classification, and the OMIM, DisGeNET, and GeneCards databases were used to investigate the gene-disease associations.
WES identified a novel homozygous GDF9 gene variant (NM_005260.6: c.275T>C; Leu92Pro) in the POI-affected sisters. PCR amplification (95°C for 5 min, followed by 28 cycles at 95°C for 30 sec, 58°C for 45 sec, 72°C for 30 sec, and 72°C for 5 min), Sanger sequencing, and subsequent segregation analysis were conducted using specifically designed forward 5′-TTTTCCTATTAGCCTTGGTTCTC-3′ and reverse 5′-CACATACCTGTTACCTGGTCTCC-3′ primers (Figures 1 and 2).
The variant was observed to be absent from the Iranome database (https://iranome.com) and Genome Aggregation Database v2.1.1 (gnomAD v2.1.1) (https://gnomad.broadinstitute.org). Based on American College of Medical Genetics and Genomics criteria, the c.275T>C variant was categorized as a variant of uncertain significance, supported by PM1, PM2, and PP3 evidence. Analyses using various in-silico tools, such as I-mutant2.0 and MutationTaster, predicted the effect of the variant (Table I). Conservation assessment was performed using ConSurf and Clustal Omega (Figure 3). Structural prediction of the region containing Leu92 was generated using the PSIPRED database (https://bioinf.cs.ucl.ac.uk) (Figure 4), and 3-dimensional molecular modeling was conducted with Chimera software (Figure 5).
2.1. Ethical Considerations
Written informed consent was obtained from both cases in this study.
3. Discussion
POI manifests through ovarian dysfunction and menopause-like symptoms, while genetic defects and autoimmune disorders serve as the main causes for its development. However, the underlying reason remains unidentified in more than half of POI cases (7).
In this study, we describe a family where 2 sisters were affected by early secondary amenorrhea before age 20. WES analysis identified a homozygous GDF9 variant (c.275T>C; Leu92Pro) in the probands, whereas their parents and grandmother were heterozygous carriers. Neither the carrier mother nor the grandmother exhibited clinical features of POI or premature menopause. Their mother and grandmother experienced menopause at ages 49 and 55, respectively. The variant was absent in population databases, and in silico analyses predicted its deleterious effects. According to the PSIPRED database, leucine at position 92 was located within an α-helix. Therefore, the Leu92Pro substitution was expected to disrupt local protein structure and α-helix formation.
GDF9 functions as an oocyte-produced growth factor that plays a crucial role in the initial stages of follicle development and in maintaining ovarian health. Different genetic variations in the GDF9 gene show association with both poor follicle development and diminished ovarian reserve, alongside cases of familial POI. Functional studies indicate that several GDF9 missense variants impair protein maturation and reduce the biological activity of conditioned media from HEK293F cells compared with the wild-type protein (8).
Initial cohort studies suggested that heterozygous GDF9 variants may cause POI in an autosomal dominant manner. Studies in French, Chinese, and Indian cohorts identified several heterozygous missense variants, including p.Ser186Tyr, p.Pro103Ser, p.Thr238Ala, p.Lys67Glu, and p.Val216Met in individuals with POI. Most variants were rare, and their pathogenicity remained uncertain. However, functional assays demonstrated the effect of missense variants, such as p.Lys67Glu and p.Pro103Ser, on protein function. In contrast, recent studies indicate that heterozygous variants have a milder effect. These studies suggested autosomal recessive inheritance of the disease due to GDF9 variants. They also claimed that heterozygous variants of this gene are associated with normal ovarian function or secondary amenorrhea at older ages (1, 8-11) (Table II).
The normal phenotypes of the mother and grandmother of our study proband further support the recessive mode of inheritance. It was observed that the clinical severity of POI is influenced by both the number of affected GDF9 alleles and the specific nature of the mutations. While a single heterozygous missense variant may play a contributory role in the onset of premature menopause, it may not necessarily lead to early menopause in all carriers. In contrast, compound heterozygous or homozygous variants that were experimentally confirmed or predicted to result in substantial impairment of protein function were more likely to cause POI or early amenorrhea.
4. Conclusion
In conclusion, we report a homozygous variant in GDF9 co-segregating with POI in affected sisters, providing support for a Mendelian gene-disease association. This finding also expands the known mutational spectrum of GDF9 and underscores its importance for genetic counseling and molecular diagnosis in families affected by POI.
Author Contributions
B. Haj Mohammad Hassani: Conceptualization, bioinformatics analysis, literature review, and article writing. N. Ghasemi: Literature review and article writing. K. Malekzadeh: Data collection, supervision, editing, and review.
Acknowledgments
The researchers sincerely appreciate both the cases and the health service staff of Shahid Mohammadi hospital, Bandar Abbas, Iran who contributed to this research. The authors declare that no funds, grants, or other support were received during the preparation of this manuscript. The authors of this article did not use artificial intelligence (AI) in any way.
Conflict of Interest
The authors declare that there is no conflict of interest.
Type of Study: Case Report |
Subject:
Reproductive Genetics
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
