
International Journal of
Reproductive Biomedicine
Tue, Jun 23, 2026
[Archive]
Volume 24, Issue 4 (April 2026)
IJRM 2026, 24(4): 337-348 |
Back to browse issues page



Ethics code: IR.UMSU.REC.1404.254
![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Fayyazi Moghaddam S, Hajshafiha M, Behroozi Lak T, Sadeghpour S, Haghtalab A. Efficacy and safety of different cetrorelix doses in the luteal phase for preventing ovarian hyperstimulation syndrome: A cross-sectional study. IJRM 2026; 24 (4) :337-348
URL: http://ijrm.ir/article-1-3798-en.html
URL: http://ijrm.ir/article-1-3798-en.html
Shiva Fayyazi Moghaddam1 

 , Masoumeh Hajshafiha *2 , Tahereh Behroozi Lak1 , Sonia Sadeghpour3 , Arian Haghtalab4
, Masoumeh Hajshafiha *2 , Tahereh Behroozi Lak1 , Sonia Sadeghpour3 , Arian Haghtalab4
, Masoumeh Hajshafiha *2 , Tahereh Behroozi Lak1 , Sonia Sadeghpour3 , Arian Haghtalab4
1- Department of Obstetrics and Gynecology, School of Medicine, Urmia University of Medical Sciences, Urmia, Iran.
2- Department of Obstetrics and Gynecology, School of Medicine, Urmia University of Medical Sciences, Urmia, Iran. ,mhajshiafha@gmail.com; hajshafiha.m@umsu.ac.ir
3- Department of Obstetrics and Gynecology, School of Medicine, Urmia University of Medical Sciences, Urmia, Iran. & Reproductive Health Research Center, Clinical Research Institute, Urmia University of Medical Sciences, Urmia, Iran.
4- School of Medicine, Urmia University of Medical Sciences, Urmia, Iran. & School of Medicine, Hamadan University of Medical Sciences, Hamadan, Iran.
2- Department of Obstetrics and Gynecology, School of Medicine, Urmia University of Medical Sciences, Urmia, Iran. ,
3- Department of Obstetrics and Gynecology, School of Medicine, Urmia University of Medical Sciences, Urmia, Iran. & Reproductive Health Research Center, Clinical Research Institute, Urmia University of Medical Sciences, Urmia, Iran.
4- School of Medicine, Urmia University of Medical Sciences, Urmia, Iran. & School of Medicine, Hamadan University of Medical Sciences, Hamadan, Iran.
Keywords: Cetrorelix, Gonadotropin-releasing hormone, Ovarian hyperstimulation syndrome, Sperm injections, Intracytoplasmic, Vascular endothelial growth factors.
Full-Text [PDF 589 kb]
(142 Downloads)
| Abstract (HTML) (205 Views)
Full-Text: (29 Views)
1. Introduction
Ovarian hyperstimulation syndrome (OHSS) is one of the potentially lethal iatrogenic complications of controlled ovarian stimulation in assisted reproductive technologies (1, 2). The phenomenon is a result of a supraphysiological ovarian response to gonadotropins, exacerbated by the delivery of human chorionic gonadotropin (hCG) for ultimate oocyte maturation. Pathophysiologically, OHSS is characterized by multiple mechanisms, notably a significant rise in vascular permeability due to pro-inflammatory cytokines and vascular endothelial growth factor (VEGF) production (3). This disorder usually presents as a mild clinical condition; however, it can become severe with several complications, leading to death. The rare but serious conditions highlight the significance of OHSS prevention for medical professionals and medicolegal risk managers (1). Specifically, among women undergoing ovulation induction and controlled ovarian stimulation, the chance of mild, moderate, and severe OHSS can be up to 30% for mild cases (4), 7% for moderate cases, and 2% for severe cases, spotlighting it as a serious risk associated with assisted reproductive technologies (5).
To decrease such risks, the gonadotropin-releasing hormone (GnRH) antagonist protocol is currently used frequently as the primary preventive strategy in clinical practice. Recent guidelines now classify this protocol as the preferred approach over traditional agonist regimens, citing high-quality evidence that it significantly lowers the incidence of OHSS without compromising live birth rates (6, 7). Also, these agents have allowed new therapies when combined with other protocols (e.g., the implementation of a GnRH agonist trigger and a "freeze-all" strategy) (3, 8). The 2 mechanisms of action that lead to efficacy, promoting very rapid luteolysis (9) and suppressing VEGF expression (10), provide a solid theoretical basis for their use. Nonetheless, studies regarding the strategy and outcomes of GnRH antagonist therapies during the luteal phase show different results.
While the doses of cetrorelix during the luteal phase are associated with a reduction in the risk of OHSS, outcomes may vary depending on the severity of the disorder. Several sources confirm that a 3-5-day protocol of 0.25 mg daily effectively reduces moderate-to-severe OHSS compared to placebos (11, 12). However, other findings indicate that the same dosage immediately after oocyte retrieval may be insufficient to prevent severe early-onset OHSS (13).
From March 2022 and January 2023, clinicians at our center observed that daily cetrorelix dosages of 0.25 mg, 0.5 mg, and even 0.75 mg were beneficial in preventing OHSS in participants at elevated risk. Subsequently, this led to the investigation of the application of more potent cetrorelix dosages. Given the substantial socioeconomic burden of infertility management (14-17) and the potential for mortality associated with moderate to severe OHSS (6), it is now a central question to determine whether these elevated doses offer improved prevention. Consequently, different therapeutic doses were systematically compared in this study to identify the optimal threshold that maximizes OHSS prevention while ensuring patient safety. This study aimed to assess the efficacy and safety of 3 distinct cetrorelix dosage regimens administered during the early luteal phase to prevent OHSS in populations at greater risk.
2. Materials and Methods
2.1. Study design and participants
In this cross-sectional study, data of 271 women aged between 19 and 44 yr who had a high-risk profile for OHSS and presented to the Infertility Center of Kosar hospital, Urmia, Iran from March 2023 to March 2025 and underwent intracytoplasmic sperm injection were extracted from their medical records.
After review, 241 of these patients met the inclusion criteria and were included in the study. A standardized checklist was used to extract baseline demographics (age, body mass index [BMI]), clinical history (duration and cause of infertility), ovarian reserve markers (anti-Müllerian hormone [AMH] levels and antral follicle count [AFC]), and cycle-specific data (number of mature follicles). Patients were retrospectively divided into 3 groups based on the prophylactic luteal-phase cetrorelix regimen they received.
Instead of using randomized or severity-based selection at admission, group allocation was based on a sequential clinical strategy that the center's clinicians gradually modified. Initially, the protocol utilized a dose of 0.5 mg twice daily (BID) (group 1, n = 101). Upon observing inadequate clinical responses, the regimen was subsequently intensified to 0.5 mg 3 times a day (TDS) (group 2, n = 38). Ultimately, to further optimize efficacy, the dose was increased to 0.75 mg TDS (group 3, n = 102), which yielded the most favorable outcomes.
Regarding the safety of the highest administered dose (0.75 mg TDS, totaling 2.25 mg/day), this regimen falls within safety margins. According to the FDA pharmacology review and the official product monograph, a standard single-dose regimen of cetrorelix for preventing a luteinizing hormone (LH) surge is 3 mg (18, 19). Furthermore, single doses of up to 120 mg have been tolerated in humans without signs of toxicity (19). The cumulative dose over the 3-day treatment period in our highest-dose group (6.75 mg) remained < 10% of the proven safe maximal human dose.
While a single 3 mg bolus could theoretically have been utilized, our center only had access to 0.25 mg cetrorelix vials. Administering a 2.25 mg bolus would have required 9 simultaneous injections, which would be overly painful for the patients. Therefore, a separate dosing strategy was employed to maximize patient compliance.
2.2. Eligibility criteria
The initial cohort for initiating prophylaxis comprised participants undergoing an intracytoplasmic sperm injection cycle with a high-risk profile for OHSS, as determined by a reproductive endocrinology and infertility subspecialist. The high-risk profile in this study w:as char:acterized by the presence of polycystic ovary syndrome (PCOS) (20) (diagnosed according to the Rotterdam criteria) or a meeting of all the following objective criteria: an AFC > 16 (21) assessed via ultrasonography; a serum AMH level surpassing 3.5 ng/ml (22); and the retrieval of > 14 oocytes during ovum pick-up (20). A final inclusion criterion was the documented receipt of prophylactic luteal-phase cetrorelix, initiated immediately on the day of oocyte retrieval (Day 0), within 2-4 hr post-procedure.
Participants from the initial high-risk cohort were excluded from the final analysis if they did not receive the prophylactic post-retrieval cetrorelix intervention or were on other OHSS-preventive prescriptions. Additional exclusion criteria included medical records that contained significant omissions of important clinical or laboratory data, as well as a lack of follow-up data within the first 2 wk after retrieval. This concluded in an ultimate cohort of 241 participants for the analysis.
2.3. Treatment protocols and intervention groups
Controlled ovarian stimulation was carried out using either a long GnRH agonist protocol or a flexible GnRH antagonist protocol, as the center's standard procedure. In the GnRH agonist protocol, Buserelin (0.5 mg, subcutaneous; CinnaGen, Iran) was started in the previous cycle's mid-luteal phase. On day 2 of the subsequent menstrual cycle, gonadotropin therapy began with follicle-stimulating hormone (75 IU; Pooyesh Darou, Iran) and human menopausal gonadotropin (75 IU follicle-stimulating hormone /75 IU LH; Daroopakhsh, Iran). Each participant's gonadotropin dosage was modified based on their age, BMI, AFC, and prior ovarian response. When the average diameter of at least 3 follicles reached ≥ 18 mm, an hCG dose of 5000-10,000 IU (Pooyesh Darou, Iran) was injected to induce oocyte maturation and LH surge. Alternatively, in the GnRH antagonist protocol, gonadotropin therapy was initiated on day 2 of the menstrual cycle, followed by daily subcutaneous injections of cetrorelix (Cetrotide, 0.25 mg; Actoverco, Iran) when the lead follicle reached approximately 14 mm in diameter, continuing until the day of hCG administration for LH surge.
Because no defined optimal dose regimen for luteal-phase cetrorelix has been established, the ultimate objective of this research was to compare 3 intensified regimens. In deciding the dosages, we hypothesized that higher, more frequent doses would produce more pronounced VEGF suppression and faster luteolysis. As a result, the low-dose regimen was developed as a substantial increase over previously studied protocols, and the high-dose regimen was chosen to test the upper limits of the dose-response relationship. For this analysis, participants were retrospectively categorized into 3 distinct intervention groups based on the dosage regimen they received: those who received 0.5 mg BID, those who received 0.5 mg TDS, and those who received 0.75 mg TDS. Given that the 3 mg single dose is one of the treatment protocols (23) and that regulatory pharmacologic data indicate that single human doses up to 120 mg are well tolerated without indications of overdosage (19), it is noteworthy that these dosages fall within the safety range.
Based on the protocol of each group (regarding frequency and dosage), injections were continued on an outpatient basis for up to 3 days. The patients’ embryos across all 3 groups were cryopreserved and utilized for transfer in subsequent cycles.
2.4. Outcome measures and OHSS classification
The primary objective of the study was to quantify the prevalence and severity (mild, moderate, severe, or critical) of early-onset OHSS within the initial 9 days following oocyte retrieval and its association with the supplied dosage. The severity of OHSS was evaluated using the classification system, which is commonly used by the American Society for Reproductive Medicine (6), and covers a range of clinical and laboratory findings. The attending subspecialist's clinical assessment of the severity of symptoms, especially uncontrollable nausea and vomiting, which indicated a significant risk of rapid progression to moderate OHSS, determined those mild OHSS cases requiring hospitalization. As a result, mild cases that could be managed as outpatients were not hospitalized and were thus omitted. Also, adverse effects and hospitalizations were documented during the follow-up.
2.5. Data collection and follow-up
Patients were informed about the symptoms of OHSS and the necessary actions to be taken. In the event of any problems during the first 72 hr post-procedure, patients visited the hospital’s emergency department, and the infertility fellows were notified. If experiencing issues within the initial 72 hr, patients would visit the infertility department to receive information regarding their developed embryos and their cryopreservation. At the same time, an abdominal ultrasound and clinical examination were performed for the patients. If there were any problems 72 hr after the procedure, patients would return to the infertility clinic, where necessary evaluations regarding the presence and severity of OHSS were conducted. At any stage of the patient visits, if symptoms indicative of OHSS were present, the necessary assessments for OHSS were carried out, the severity of the OHSS was determined, and the patients were hospitalized if admission was required.
The baseline data gathered included demographic data (age, BMI), clinical history (type and duration of infertility, and primary diagnosis), and ovarian reserve markers (AMH and AFC). Cycle-specific data were gathered regarding the quantity of mature follicles at the point of trigger. The primary outcome was the rate and severity of OHSS, and the clinical outcome of hospitalization was also documented during the 2 wk post-oocyte retrieval in all participants. To maintain data integrity, a second investigator manually verified a random 10% sample of the abstracted data and resolved any differences by consensus.
2.6. Bias mitigation
Potential sources of bias were systematically identified and rectified, whereas selection bias was alleviated by enrolling all eligible participants meeting the high-risk criteria over the study period. To address information bias, a standardized data abstraction form was used, along with strict exclusion criteria for records with ambiguous or incomplete data. Finally, the statistical analysis used multivariable regression models to account for confounding caused by baseline variables known to influence OHSS risk.
2.7. Ethical Considerations
This study was approved by the ethics committee of Urmia University of Medical Sciences, Urmia, Iran (Code: IR.UMSU.REC.1404.254), adhering to the Declaration of Helsinki. The committee waived the requirement for specific participant permission due to the study's retrospective design and the utilization of de-identified data. All participants’ data were thoroughly anonymized before the study, so as to guarantee participant confidentiality.
2.8. Statistical Analysis
Continuous variables were presented as median (interquartile range) after normality assessment done via the Shapiro-Wilk test, while categorical variables were reported as frequencies and percentages. The Kruskal-Wallis test was employed to compare continuous variables across dosage groups, and the Pearson Chi-square test was utilized for categorical comparisons and OHSS severity distribution. Multivariable binary logistic regression determined independent predictors of hospitalization, presenting results as adjusted odds ratios (AORs) with 95% confidence intervals (CIs), and sensitivity analyses were performed to verify robustness. Statistical analyses were conducted using IBM SPSS Statistics version 27.0 (IBM Corp., Armonk, NY), with a p < 0.05 considered statistically significant.
3. Results
3.1. Participant characteristics
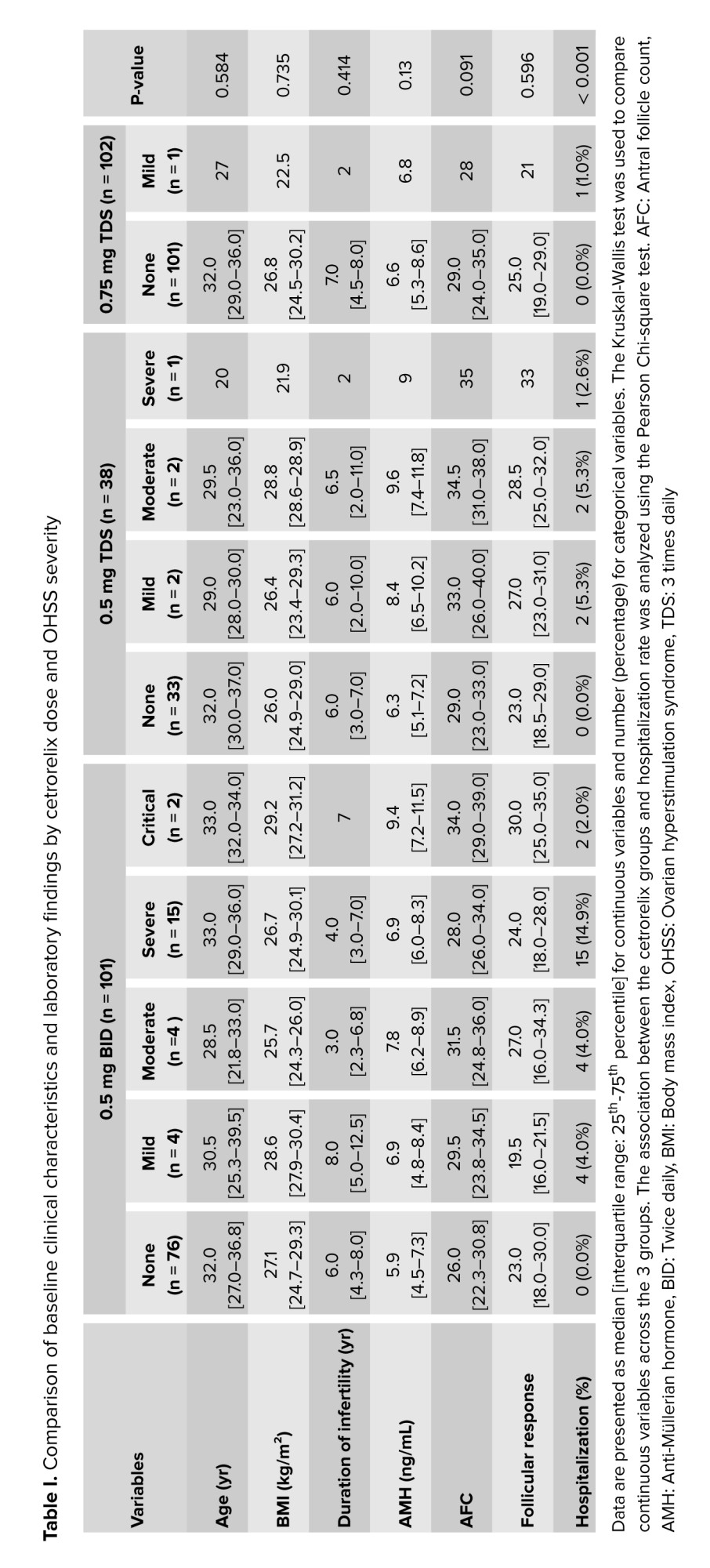
A total of 241 participants with a higher risk of OHSS were incorporated in the final analysis. Participants were allocated to one of 3 luteal-phase cetrorelix protocols of 0.5 mg BID (n = 101), 0.5 mg TDS (n = 38), or 0.75 mg TDS (n = 102). The baseline demographic and clinical features were equivalent among the 3 regimen groups. No statistically significant differences were observed in the overall median age, BMI, infertility duration, AMH, AFC, or follicular response among the groups (p > 0.05 for all comparisons) (Table I). Furthermore, the distribution of infertility diagnoses was also similar across the 3 treatment arms (p = 0.388), with PCOS being the most common diagnosis in all groups (Table II).
3.2. Efficacy of cetrorelix doses in preventing OHSS
The preventative efficacy against OHSS was clearly dose-dependent; the overall incidence of any grade of OHSS was 24.8% in the 0.5 mg BID group, 13.2% in the 0.5 mg TDS group, and much lower (1.0% in the 0.75 mg TDS group) (Table I; χ²(2) = 25.590, p < 0.001). This effect was more pronounced with clinically significant OHSS (moderate, severe, and critical), with incidences of 20.8% with the 0.5 mg BID dose and 7.9% with the 0.5 mg TDS dose. Notably, no participants (0%) in the highest dose (0.75 mg TDS) group developed moderate or higher severity OHSS. Also, the dosage group showed a significant correlation with severity (χ² (8) = 31.022, p < 0.001).
3.3. Predictors of hospitalization
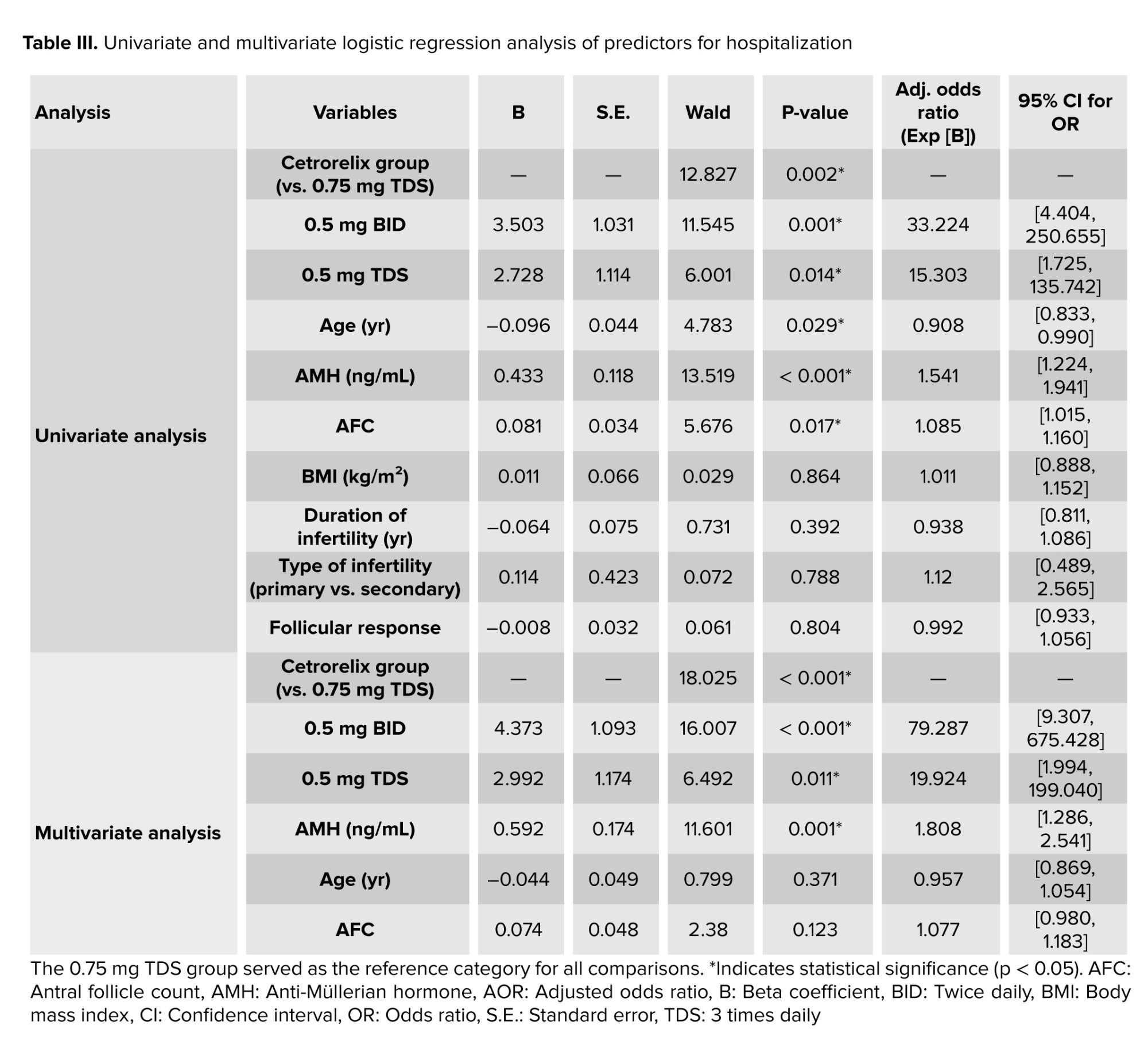
Univariate and multivariable logistic regression models were employed to identify risk factors for hospitalization (Table III). The initial univariate analysis revealed cetrorelix group (p = 0.002), age (p = 0.029), AMH (p < 0.001), and AFC (p = 0.017) as significant predictors. The finalized multivariable model demonstrated statistical significance (p < 0.001) and explained 42.2% of hospitalization variation. After adjusting for confounders, only cetrorelix dosage and AMH level remained as strong, independent predictors. Compared to the 0.75 mg TDS reference group, the odds of hospitalization for participants in the 0.5 mg BID group were over 79 times higher (Adjusted OR = 79.29; 95% CI [9.31, 675.43]; p < 0.001). The odds for the 0.5 mg TDS group were nearly 20 times higher (Adjusted OR = 19.92; 95% CI [1.99, 199.04]; p = 0.011). Higher AMH also remained a significant risk factor, with each one ng/mL increase associated with an 81% increase in the odds of hospitalization (AOR = 1.81; 95% CI [1.29, 2.54]; p = 0.001). In the final model, age (p = 0.371) and AFC (p = 0.123) were no longer statistically significant.
3.4. Side effects
The cetrorelix protocols were generally well-tolerated by the study population. The most common side effect was minor redness at the site of injection in the first place, and then a transient rise in liver function (LFTs) tests. The redness at the site of injection was reported in 26 of the 241 participants (10.8%). It is noteworthy that none of the participants who experienced redness at the site of injection required hospitalization for OHSS. A transient rise in LFTs was observed in 8 participants, including 2 participants who were hospitalized for OHSS. However, all the elevations were minor and did not exceed 2 times the upper limit of the normal range. No other side effects were observed during the study.

4. Discussion
This study examined the effectiveness of 3 different luteal-phase cetrorelix regimens in the prevention of OHSS among high-risk participants. Our main finding was that a 0.75 mg TDS protocol was much more effective than low-dose regimens (0.5 mg BID and 0.5 mg TDS) in terms of the overall incidence and severity of OHSS. No cases of critical, severe, or even moderate OHSS were noted in the highest-dose group, and the adjusted likelihood of hospitalization was almost 20-fold lower than in the medium-dose group and almost 79-fold lower than in the lowest-dose group.
Our findings align with previous evidence supporting the utilization of GnRH antagonists for the prevention of OHSS during the luteal phase.
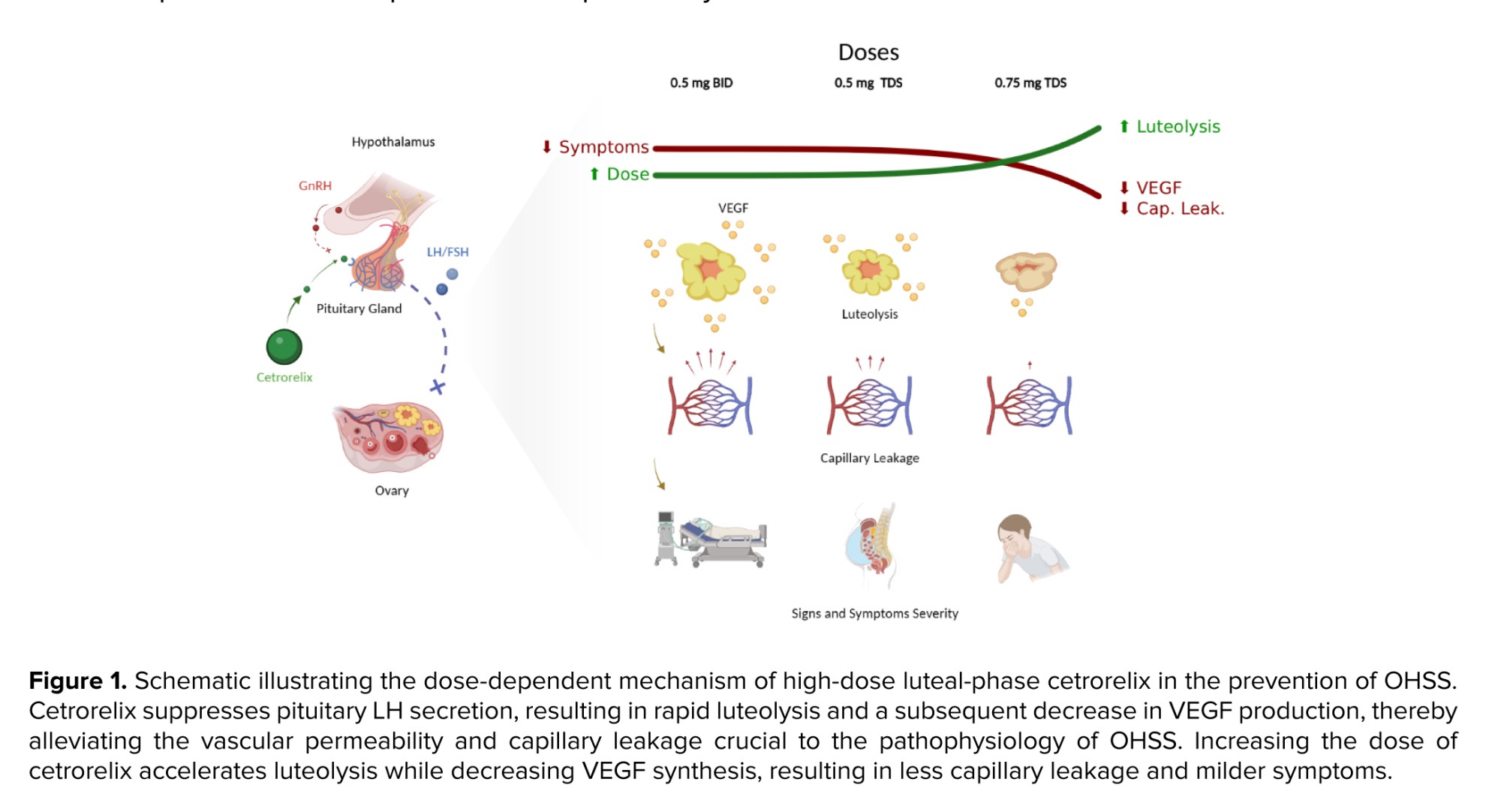
While comparing to other studies, several studies have shown that a standard luteal-phase dose of cetrorelix (0.25 mg daily) markedly minimized the likelihood of moderate-to-severe OHSS in contrast with controls (11, 12). Our study provides extended findings and enhanced evidence for a dose-dependent effect, as our lowest-dose group (1.0 mg/day) had a total daily dose significantly higher than previous studies. Additionally, our highest-dose group (2.25 mg/day) enhanced the protective effects. This new finding indicates that aggressive suppression of the VEGF pathway and further enhanced luteolysis may optimize outcomes in the highest-risk participants (Figure 1).
Notably, our results highlights that this treatment is dose-dependent, especially when compared to an opposite finding from a study which found that moderate or severe OHSS was not significantly affected by 0.25 mg cetrorelix (13). This lack of effect probably means that the protective mechanisms seen in our higher-dose protocols were not triggered by their dosage. In fact, variations in stimulation protocols, participant populations, and probably dosage could all contribute to this discrepancy.
However, it is notable that several other studies provided a supporting framework for our findings, which helped us to develop the dose-dependency hypothesis (24, 25). For example, in 2017, a randomized controlled trial was performed in which participants received 0.25 mg BID of an antagonist the day before the hCG trigger, which eliminated moderate/severe OHSS (0% vs. 12.37% in the standard-dose group) (25). Also, another study showed that doubling the antagonist dose effectively mitigated estradiol levels and did not result in OHSS development in high-responder oocyte donors (24). Although the doses were different from our study, the concept was the same, and that is, higher doses of a GnRH antagonist provide more effective preventative effects by decreasing VEGF production and its effects on capillary leakage and leading to OHSS, even though there was an essential variation in the timing of administration.
4.1. Strengths and Limitations
Our study's primary finding was the significant baseline difference in OHSS risk markers between the 3 groups. The high-dose participants showed higher AMH and AFC levels, indicating an increased risk of OHSS. This raises the likelihood of confounding by indication, with physicians providing aggressive therapy for higher-risk participants. Furthermore, the highest-risk sample had the best overall outcomes, supporting the efficacy of high-dose regimens. The multivariable logistic regression study, which controlled AMH and other factors, revealed that dosage was an independent and strong predictor of positive outcomes. However, this study had several limitations.
First, because of its retrospective, non-randomized design, this study fails to demonstrate a cause-and-effect link; nonetheless, numerous steps were made to reduce this natural vulnerability. Enrolling eligible participants in a time order reduced selection bias, whereas multivariable regression analysis controlled for confounding variables such as baseline AMH and AFC. However, unmeasured confounding variables may cause residual bias.
Second, the study was conducted at a single Iranian center, which may restrict its generalizability.
Third, the wide CIs were observed in the multivariable regression analysis, particularly regarding the risk of hospitalization in the low-dose groups (e.g., 95% CI: 9.31-675.43). This is attributable to the limited sample size in the 0.5 mg TDS subgroup (n = 38) and the zero-event outcome in the reference group (0.75 mg TDS). Although this confirms the high efficacy of the maximal dose, the resulting sparse data relative to the number of covariates contributes to lower precision in the point estimates for the odds ratios.
Finally, while this study reveals a positive clinical outcome, it does not provide any data to evaluate the potential underlying molecular factors. Key biomarker tests, such as serum VEGF to validate enhanced vascular permeability or progesterone/estradiol levels to establish the depth and speed of luteolysis, were not used. Molecular analysis would be required to investigate the biological causality of the dose-dependent effects described.
5. Conclusion
While there are certain limitations to the study, the data supports a firm conclusion that an intensive, high-dose luteal-phase regimen of cetrorelix (0.75 mg TDS) is effective in preventing moderate, severe, and critical OHSS in a population at increased risk. The high-dose regimen has shown superior effectiveness compared to the lower dose regimens, 0.5 mg BID and 0.5 mg TDS, even after adjusting for baseline risk factors. This result suggests a dose-dependent relationship in the luteal-phase protective effect of cetrorelix. While our findings are very promising, they need to be validated in a larger, prospective randomized controlled trial for it to become a standard of care for risk-reducing OHSS treatment for participants at very high risk.
Data Availability
Data would be available upon online request from the corresponding author.
Author Contributions
Sh. Fayyazi Moghaddam and M. Hajshafiha: Conceptualized the study, designed the methodology, administered the project, performed formal analysis, and wrote the original draft. T. Behroozi Lak: Participated in data curation, provided resources, and validated the results. S. Sadeghpour: Contributed to supervision and validation. A. Haghtalab: Performed the formal analysis, visualization, and helped draft the manuscript. All authors participated in the review and editing process and read and approved the final manuscript.
Acknowledgments
The authors acknowledge the staff of Kowsar hospital of Urmia, Urmia, Iran, for their unlimited support during the study. The authors declare that no funds, grants, or other support were received during the preparation of this manuscript. We also extend our gratitude to the developers of Grammarly, which was used for checking grammar in the manuscript.
Conflict of Interest
The authors declare that there is no conflict of interest.
Ovarian hyperstimulation syndrome (OHSS) is one of the potentially lethal iatrogenic complications of controlled ovarian stimulation in assisted reproductive technologies (1, 2). The phenomenon is a result of a supraphysiological ovarian response to gonadotropins, exacerbated by the delivery of human chorionic gonadotropin (hCG) for ultimate oocyte maturation. Pathophysiologically, OHSS is characterized by multiple mechanisms, notably a significant rise in vascular permeability due to pro-inflammatory cytokines and vascular endothelial growth factor (VEGF) production (3). This disorder usually presents as a mild clinical condition; however, it can become severe with several complications, leading to death. The rare but serious conditions highlight the significance of OHSS prevention for medical professionals and medicolegal risk managers (1). Specifically, among women undergoing ovulation induction and controlled ovarian stimulation, the chance of mild, moderate, and severe OHSS can be up to 30% for mild cases (4), 7% for moderate cases, and 2% for severe cases, spotlighting it as a serious risk associated with assisted reproductive technologies (5).
To decrease such risks, the gonadotropin-releasing hormone (GnRH) antagonist protocol is currently used frequently as the primary preventive strategy in clinical practice. Recent guidelines now classify this protocol as the preferred approach over traditional agonist regimens, citing high-quality evidence that it significantly lowers the incidence of OHSS without compromising live birth rates (6, 7). Also, these agents have allowed new therapies when combined with other protocols (e.g., the implementation of a GnRH agonist trigger and a "freeze-all" strategy) (3, 8). The 2 mechanisms of action that lead to efficacy, promoting very rapid luteolysis (9) and suppressing VEGF expression (10), provide a solid theoretical basis for their use. Nonetheless, studies regarding the strategy and outcomes of GnRH antagonist therapies during the luteal phase show different results.
While the doses of cetrorelix during the luteal phase are associated with a reduction in the risk of OHSS, outcomes may vary depending on the severity of the disorder. Several sources confirm that a 3-5-day protocol of 0.25 mg daily effectively reduces moderate-to-severe OHSS compared to placebos (11, 12). However, other findings indicate that the same dosage immediately after oocyte retrieval may be insufficient to prevent severe early-onset OHSS (13).
From March 2022 and January 2023, clinicians at our center observed that daily cetrorelix dosages of 0.25 mg, 0.5 mg, and even 0.75 mg were beneficial in preventing OHSS in participants at elevated risk. Subsequently, this led to the investigation of the application of more potent cetrorelix dosages. Given the substantial socioeconomic burden of infertility management (14-17) and the potential for mortality associated with moderate to severe OHSS (6), it is now a central question to determine whether these elevated doses offer improved prevention. Consequently, different therapeutic doses were systematically compared in this study to identify the optimal threshold that maximizes OHSS prevention while ensuring patient safety. This study aimed to assess the efficacy and safety of 3 distinct cetrorelix dosage regimens administered during the early luteal phase to prevent OHSS in populations at greater risk.
2. Materials and Methods
2.1. Study design and participants
In this cross-sectional study, data of 271 women aged between 19 and 44 yr who had a high-risk profile for OHSS and presented to the Infertility Center of Kosar hospital, Urmia, Iran from March 2023 to March 2025 and underwent intracytoplasmic sperm injection were extracted from their medical records.
After review, 241 of these patients met the inclusion criteria and were included in the study. A standardized checklist was used to extract baseline demographics (age, body mass index [BMI]), clinical history (duration and cause of infertility), ovarian reserve markers (anti-Müllerian hormone [AMH] levels and antral follicle count [AFC]), and cycle-specific data (number of mature follicles). Patients were retrospectively divided into 3 groups based on the prophylactic luteal-phase cetrorelix regimen they received.
Instead of using randomized or severity-based selection at admission, group allocation was based on a sequential clinical strategy that the center's clinicians gradually modified. Initially, the protocol utilized a dose of 0.5 mg twice daily (BID) (group 1, n = 101). Upon observing inadequate clinical responses, the regimen was subsequently intensified to 0.5 mg 3 times a day (TDS) (group 2, n = 38). Ultimately, to further optimize efficacy, the dose was increased to 0.75 mg TDS (group 3, n = 102), which yielded the most favorable outcomes.
Regarding the safety of the highest administered dose (0.75 mg TDS, totaling 2.25 mg/day), this regimen falls within safety margins. According to the FDA pharmacology review and the official product monograph, a standard single-dose regimen of cetrorelix for preventing a luteinizing hormone (LH) surge is 3 mg (18, 19). Furthermore, single doses of up to 120 mg have been tolerated in humans without signs of toxicity (19). The cumulative dose over the 3-day treatment period in our highest-dose group (6.75 mg) remained < 10% of the proven safe maximal human dose.
While a single 3 mg bolus could theoretically have been utilized, our center only had access to 0.25 mg cetrorelix vials. Administering a 2.25 mg bolus would have required 9 simultaneous injections, which would be overly painful for the patients. Therefore, a separate dosing strategy was employed to maximize patient compliance.
2.2. Eligibility criteria
The initial cohort for initiating prophylaxis comprised participants undergoing an intracytoplasmic sperm injection cycle with a high-risk profile for OHSS, as determined by a reproductive endocrinology and infertility subspecialist. The high-risk profile in this study w:as char:acterized by the presence of polycystic ovary syndrome (PCOS) (20) (diagnosed according to the Rotterdam criteria) or a meeting of all the following objective criteria: an AFC > 16 (21) assessed via ultrasonography; a serum AMH level surpassing 3.5 ng/ml (22); and the retrieval of > 14 oocytes during ovum pick-up (20). A final inclusion criterion was the documented receipt of prophylactic luteal-phase cetrorelix, initiated immediately on the day of oocyte retrieval (Day 0), within 2-4 hr post-procedure.
Participants from the initial high-risk cohort were excluded from the final analysis if they did not receive the prophylactic post-retrieval cetrorelix intervention or were on other OHSS-preventive prescriptions. Additional exclusion criteria included medical records that contained significant omissions of important clinical or laboratory data, as well as a lack of follow-up data within the first 2 wk after retrieval. This concluded in an ultimate cohort of 241 participants for the analysis.
2.3. Treatment protocols and intervention groups
Controlled ovarian stimulation was carried out using either a long GnRH agonist protocol or a flexible GnRH antagonist protocol, as the center's standard procedure. In the GnRH agonist protocol, Buserelin (0.5 mg, subcutaneous; CinnaGen, Iran) was started in the previous cycle's mid-luteal phase. On day 2 of the subsequent menstrual cycle, gonadotropin therapy began with follicle-stimulating hormone (75 IU; Pooyesh Darou, Iran) and human menopausal gonadotropin (75 IU follicle-stimulating hormone /75 IU LH; Daroopakhsh, Iran). Each participant's gonadotropin dosage was modified based on their age, BMI, AFC, and prior ovarian response. When the average diameter of at least 3 follicles reached ≥ 18 mm, an hCG dose of 5000-10,000 IU (Pooyesh Darou, Iran) was injected to induce oocyte maturation and LH surge. Alternatively, in the GnRH antagonist protocol, gonadotropin therapy was initiated on day 2 of the menstrual cycle, followed by daily subcutaneous injections of cetrorelix (Cetrotide, 0.25 mg; Actoverco, Iran) when the lead follicle reached approximately 14 mm in diameter, continuing until the day of hCG administration for LH surge.
Because no defined optimal dose regimen for luteal-phase cetrorelix has been established, the ultimate objective of this research was to compare 3 intensified regimens. In deciding the dosages, we hypothesized that higher, more frequent doses would produce more pronounced VEGF suppression and faster luteolysis. As a result, the low-dose regimen was developed as a substantial increase over previously studied protocols, and the high-dose regimen was chosen to test the upper limits of the dose-response relationship. For this analysis, participants were retrospectively categorized into 3 distinct intervention groups based on the dosage regimen they received: those who received 0.5 mg BID, those who received 0.5 mg TDS, and those who received 0.75 mg TDS. Given that the 3 mg single dose is one of the treatment protocols (23) and that regulatory pharmacologic data indicate that single human doses up to 120 mg are well tolerated without indications of overdosage (19), it is noteworthy that these dosages fall within the safety range.
Based on the protocol of each group (regarding frequency and dosage), injections were continued on an outpatient basis for up to 3 days. The patients’ embryos across all 3 groups were cryopreserved and utilized for transfer in subsequent cycles.
2.4. Outcome measures and OHSS classification
The primary objective of the study was to quantify the prevalence and severity (mild, moderate, severe, or critical) of early-onset OHSS within the initial 9 days following oocyte retrieval and its association with the supplied dosage. The severity of OHSS was evaluated using the classification system, which is commonly used by the American Society for Reproductive Medicine (6), and covers a range of clinical and laboratory findings. The attending subspecialist's clinical assessment of the severity of symptoms, especially uncontrollable nausea and vomiting, which indicated a significant risk of rapid progression to moderate OHSS, determined those mild OHSS cases requiring hospitalization. As a result, mild cases that could be managed as outpatients were not hospitalized and were thus omitted. Also, adverse effects and hospitalizations were documented during the follow-up.
2.5. Data collection and follow-up
Patients were informed about the symptoms of OHSS and the necessary actions to be taken. In the event of any problems during the first 72 hr post-procedure, patients visited the hospital’s emergency department, and the infertility fellows were notified. If experiencing issues within the initial 72 hr, patients would visit the infertility department to receive information regarding their developed embryos and their cryopreservation. At the same time, an abdominal ultrasound and clinical examination were performed for the patients. If there were any problems 72 hr after the procedure, patients would return to the infertility clinic, where necessary evaluations regarding the presence and severity of OHSS were conducted. At any stage of the patient visits, if symptoms indicative of OHSS were present, the necessary assessments for OHSS were carried out, the severity of the OHSS was determined, and the patients were hospitalized if admission was required.
The baseline data gathered included demographic data (age, BMI), clinical history (type and duration of infertility, and primary diagnosis), and ovarian reserve markers (AMH and AFC). Cycle-specific data were gathered regarding the quantity of mature follicles at the point of trigger. The primary outcome was the rate and severity of OHSS, and the clinical outcome of hospitalization was also documented during the 2 wk post-oocyte retrieval in all participants. To maintain data integrity, a second investigator manually verified a random 10% sample of the abstracted data and resolved any differences by consensus.
2.6. Bias mitigation
Potential sources of bias were systematically identified and rectified, whereas selection bias was alleviated by enrolling all eligible participants meeting the high-risk criteria over the study period. To address information bias, a standardized data abstraction form was used, along with strict exclusion criteria for records with ambiguous or incomplete data. Finally, the statistical analysis used multivariable regression models to account for confounding caused by baseline variables known to influence OHSS risk.
2.7. Ethical Considerations
This study was approved by the ethics committee of Urmia University of Medical Sciences, Urmia, Iran (Code: IR.UMSU.REC.1404.254), adhering to the Declaration of Helsinki. The committee waived the requirement for specific participant permission due to the study's retrospective design and the utilization of de-identified data. All participants’ data were thoroughly anonymized before the study, so as to guarantee participant confidentiality.
2.8. Statistical Analysis
Continuous variables were presented as median (interquartile range) after normality assessment done via the Shapiro-Wilk test, while categorical variables were reported as frequencies and percentages. The Kruskal-Wallis test was employed to compare continuous variables across dosage groups, and the Pearson Chi-square test was utilized for categorical comparisons and OHSS severity distribution. Multivariable binary logistic regression determined independent predictors of hospitalization, presenting results as adjusted odds ratios (AORs) with 95% confidence intervals (CIs), and sensitivity analyses were performed to verify robustness. Statistical analyses were conducted using IBM SPSS Statistics version 27.0 (IBM Corp., Armonk, NY), with a p < 0.05 considered statistically significant.
3. Results
3.1. Participant characteristics
A total of 241 participants with a higher risk of OHSS were incorporated in the final analysis. Participants were allocated to one of 3 luteal-phase cetrorelix protocols of 0.5 mg BID (n = 101), 0.5 mg TDS (n = 38), or 0.75 mg TDS (n = 102). The baseline demographic and clinical features were equivalent among the 3 regimen groups. No statistically significant differences were observed in the overall median age, BMI, infertility duration, AMH, AFC, or follicular response among the groups (p > 0.05 for all comparisons) (Table I). Furthermore, the distribution of infertility diagnoses was also similar across the 3 treatment arms (p = 0.388), with PCOS being the most common diagnosis in all groups (Table II).
3.2. Efficacy of cetrorelix doses in preventing OHSS
The preventative efficacy against OHSS was clearly dose-dependent; the overall incidence of any grade of OHSS was 24.8% in the 0.5 mg BID group, 13.2% in the 0.5 mg TDS group, and much lower (1.0% in the 0.75 mg TDS group) (Table I; χ²(2) = 25.590, p < 0.001). This effect was more pronounced with clinically significant OHSS (moderate, severe, and critical), with incidences of 20.8% with the 0.5 mg BID dose and 7.9% with the 0.5 mg TDS dose. Notably, no participants (0%) in the highest dose (0.75 mg TDS) group developed moderate or higher severity OHSS. Also, the dosage group showed a significant correlation with severity (χ² (8) = 31.022, p < 0.001).
3.3. Predictors of hospitalization
Univariate and multivariable logistic regression models were employed to identify risk factors for hospitalization (Table III). The initial univariate analysis revealed cetrorelix group (p = 0.002), age (p = 0.029), AMH (p < 0.001), and AFC (p = 0.017) as significant predictors. The finalized multivariable model demonstrated statistical significance (p < 0.001) and explained 42.2% of hospitalization variation. After adjusting for confounders, only cetrorelix dosage and AMH level remained as strong, independent predictors. Compared to the 0.75 mg TDS reference group, the odds of hospitalization for participants in the 0.5 mg BID group were over 79 times higher (Adjusted OR = 79.29; 95% CI [9.31, 675.43]; p < 0.001). The odds for the 0.5 mg TDS group were nearly 20 times higher (Adjusted OR = 19.92; 95% CI [1.99, 199.04]; p = 0.011). Higher AMH also remained a significant risk factor, with each one ng/mL increase associated with an 81% increase in the odds of hospitalization (AOR = 1.81; 95% CI [1.29, 2.54]; p = 0.001). In the final model, age (p = 0.371) and AFC (p = 0.123) were no longer statistically significant.
3.4. Side effects
The cetrorelix protocols were generally well-tolerated by the study population. The most common side effect was minor redness at the site of injection in the first place, and then a transient rise in liver function (LFTs) tests. The redness at the site of injection was reported in 26 of the 241 participants (10.8%). It is noteworthy that none of the participants who experienced redness at the site of injection required hospitalization for OHSS. A transient rise in LFTs was observed in 8 participants, including 2 participants who were hospitalized for OHSS. However, all the elevations were minor and did not exceed 2 times the upper limit of the normal range. No other side effects were observed during the study.
4. Discussion
This study examined the effectiveness of 3 different luteal-phase cetrorelix regimens in the prevention of OHSS among high-risk participants. Our main finding was that a 0.75 mg TDS protocol was much more effective than low-dose regimens (0.5 mg BID and 0.5 mg TDS) in terms of the overall incidence and severity of OHSS. No cases of critical, severe, or even moderate OHSS were noted in the highest-dose group, and the adjusted likelihood of hospitalization was almost 20-fold lower than in the medium-dose group and almost 79-fold lower than in the lowest-dose group.
Our findings align with previous evidence supporting the utilization of GnRH antagonists for the prevention of OHSS during the luteal phase.
While comparing to other studies, several studies have shown that a standard luteal-phase dose of cetrorelix (0.25 mg daily) markedly minimized the likelihood of moderate-to-severe OHSS in contrast with controls (11, 12). Our study provides extended findings and enhanced evidence for a dose-dependent effect, as our lowest-dose group (1.0 mg/day) had a total daily dose significantly higher than previous studies. Additionally, our highest-dose group (2.25 mg/day) enhanced the protective effects. This new finding indicates that aggressive suppression of the VEGF pathway and further enhanced luteolysis may optimize outcomes in the highest-risk participants (Figure 1).
Notably, our results highlights that this treatment is dose-dependent, especially when compared to an opposite finding from a study which found that moderate or severe OHSS was not significantly affected by 0.25 mg cetrorelix (13). This lack of effect probably means that the protective mechanisms seen in our higher-dose protocols were not triggered by their dosage. In fact, variations in stimulation protocols, participant populations, and probably dosage could all contribute to this discrepancy.
However, it is notable that several other studies provided a supporting framework for our findings, which helped us to develop the dose-dependency hypothesis (24, 25). For example, in 2017, a randomized controlled trial was performed in which participants received 0.25 mg BID of an antagonist the day before the hCG trigger, which eliminated moderate/severe OHSS (0% vs. 12.37% in the standard-dose group) (25). Also, another study showed that doubling the antagonist dose effectively mitigated estradiol levels and did not result in OHSS development in high-responder oocyte donors (24). Although the doses were different from our study, the concept was the same, and that is, higher doses of a GnRH antagonist provide more effective preventative effects by decreasing VEGF production and its effects on capillary leakage and leading to OHSS, even though there was an essential variation in the timing of administration.
4.1. Strengths and Limitations
Our study's primary finding was the significant baseline difference in OHSS risk markers between the 3 groups. The high-dose participants showed higher AMH and AFC levels, indicating an increased risk of OHSS. This raises the likelihood of confounding by indication, with physicians providing aggressive therapy for higher-risk participants. Furthermore, the highest-risk sample had the best overall outcomes, supporting the efficacy of high-dose regimens. The multivariable logistic regression study, which controlled AMH and other factors, revealed that dosage was an independent and strong predictor of positive outcomes. However, this study had several limitations.
First, because of its retrospective, non-randomized design, this study fails to demonstrate a cause-and-effect link; nonetheless, numerous steps were made to reduce this natural vulnerability. Enrolling eligible participants in a time order reduced selection bias, whereas multivariable regression analysis controlled for confounding variables such as baseline AMH and AFC. However, unmeasured confounding variables may cause residual bias.
Second, the study was conducted at a single Iranian center, which may restrict its generalizability.
Third, the wide CIs were observed in the multivariable regression analysis, particularly regarding the risk of hospitalization in the low-dose groups (e.g., 95% CI: 9.31-675.43). This is attributable to the limited sample size in the 0.5 mg TDS subgroup (n = 38) and the zero-event outcome in the reference group (0.75 mg TDS). Although this confirms the high efficacy of the maximal dose, the resulting sparse data relative to the number of covariates contributes to lower precision in the point estimates for the odds ratios.
Finally, while this study reveals a positive clinical outcome, it does not provide any data to evaluate the potential underlying molecular factors. Key biomarker tests, such as serum VEGF to validate enhanced vascular permeability or progesterone/estradiol levels to establish the depth and speed of luteolysis, were not used. Molecular analysis would be required to investigate the biological causality of the dose-dependent effects described.
5. Conclusion
While there are certain limitations to the study, the data supports a firm conclusion that an intensive, high-dose luteal-phase regimen of cetrorelix (0.75 mg TDS) is effective in preventing moderate, severe, and critical OHSS in a population at increased risk. The high-dose regimen has shown superior effectiveness compared to the lower dose regimens, 0.5 mg BID and 0.5 mg TDS, even after adjusting for baseline risk factors. This result suggests a dose-dependent relationship in the luteal-phase protective effect of cetrorelix. While our findings are very promising, they need to be validated in a larger, prospective randomized controlled trial for it to become a standard of care for risk-reducing OHSS treatment for participants at very high risk.
Data Availability
Data would be available upon online request from the corresponding author.
Author Contributions
Sh. Fayyazi Moghaddam and M. Hajshafiha: Conceptualized the study, designed the methodology, administered the project, performed formal analysis, and wrote the original draft. T. Behroozi Lak: Participated in data curation, provided resources, and validated the results. S. Sadeghpour: Contributed to supervision and validation. A. Haghtalab: Performed the formal analysis, visualization, and helped draft the manuscript. All authors participated in the review and editing process and read and approved the final manuscript.
Acknowledgments
The authors acknowledge the staff of Kowsar hospital of Urmia, Urmia, Iran, for their unlimited support during the study. The authors declare that no funds, grants, or other support were received during the preparation of this manuscript. We also extend our gratitude to the developers of Grammarly, which was used for checking grammar in the manuscript.
Conflict of Interest
The authors declare that there is no conflict of interest.
Type of Study: Original Article |
Subject:
Assisted Reproductive Technologies
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
